Дизостоз |
||
 |
 |
|
Оглавление
|
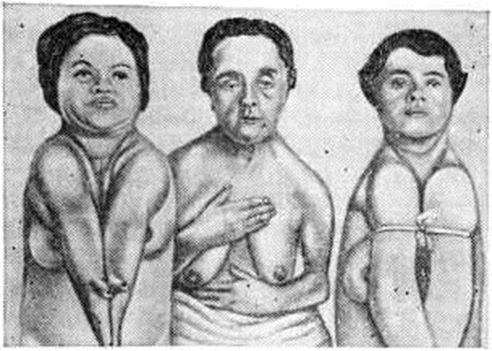
ДизостозДизостоз (dysostosis; греческий dys + osteon кость + osis) — нарушение развития костей, лежащее в основе врождённых наследственных семейных заболеваний костной системы. Чаще всего возникают аномалии развития костей черепа в сочетании с другими симптомами, однако встречаются множественные и генерализованные поражения костей скелета. Термин «дизостоз» применяют к генерализованным поражениям скелета — хондродистрофии (смотри полный свод знаний), гаргоилизму (смотри полный свод знаний), остеогенезу несовершенному (смотри полный свод знаний) и так далее Важнейшие разновидности Дизостоз: ключично-черепной, черепно-лицевой, челюстно-лицевой и челюстно-черепной. Ключично-черепной дизостоз (синдром Шейтхауэра — Мари — Сентона) характеризуется гипоплазией покровных костей черепа в сочетании с полным или частичным недоразвитием одной или обеих ключиц, то есть нарушением развития так называемый мембранозных костей. Для Дизостоз этого вида характерно незаращение или позднее заращение черепных швов и родничков, брахицефалия (смотри полный свод знаний) с преобладанием расширения свода черепа в латеральных направлениях, выдающийся лоб, гипоплазия лицевых костей, главным образом верхней челюсти, обусловливающая псевдопрогению (кажущееся увеличение нижней челюсти). Нарушение развития челюстей сопровождается запаздыванием прорезывания зубов. Отсутствие ключиц или частичное недоразвитие их с дефектом внутренних, средних или наружных частей ведёт к увеличению подвижности плечевого пояса, а при полном отсутствии их — к полному соприкосновению плеч (рисунок 1).
Описанные изменения часто сопровождаются деформациями позвоночника, костей верхних и нижних конечностей, стоп, тазовых костей. Аномалия наследуется по рецессивному и доминантному типу, может быть семейной. |
|||||||||||||
При ключично-черепном дизостозе рентгенологически выявляются многочисленные изменения со стороны скелета, однако наиболее характерны изменения ключиц и костей черепа. Дефекты ключиц чаще симметричны и могут быть разных размеров: от небольших до полного отсутствия ключиц. Чаще же всего отсутствует акромиальный конец ключицы. Свободный конец оставшейся части закруглён, покрыт замыкающей костной пластинкой и связан плотным фиброзным тяжем с акромиальным отростком лопатки. По ходу фиброзного тяжа иногда обнаруживаются костные включения.
При рентгенологическое исследовании черепа определяется брахицефалия: мозговой череп увеличен в поперечнике и уменьшен в передне-заднем размере. Основание черепа укорочено в поперечном направлении и несколько удлинено в продольном. Кости свода, особенно лобная, истончены и как бы раздуты, значительно выдаваясь в стороны. Передний родничок остаётся незаращённым. В местах перекрёста швов могут наблюдаться и дополнительные роднички или дополнительные костные включения в самих швах. Кости лицевого черепа малы, верхнечелюстные пазухи недоразвиты. Размеры нижней челюсти не изменены. Обнаруживаются аномалии прикуса, расположения, формы и сроков прорезывания зубов.
При исследовании скелета туловища и конечностей могут быть обнаружены отклонения в развитии ряда костей: уменьшенные размеры лопаток, крестца, костей таза с отсутствием слияния между собой лобковых, седалищных и подвздошных костей и недоразвитием лобкового симфиза; недоразвитие проксимальных отделов бёдер с варусной деформацией их; укорочение или отсутствие ногтевых бугристостей у концевых фаланг пальцев кистей и стоп; незаращение дужек позвонков.
При множественном поражении скелета наличие характерных изменений ключиц делает рентгенологическое диагноз достоверным.
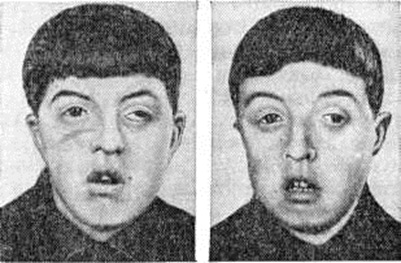
Черепно-лицевой дизостоз (синдром Крузона, гипертелоризм) — недоразвитие костей черепа, мозга и верхней челюсти в сочетании с преждевременным закрытием черепных швов, экзофтальмом (смотри полный свод знаний), косоглазием (смотри полный свод знаний), нистагмом (смотри полный свод знаний), расстройством зрения. Лоб в области переносицы бугрист, глаза широко расставлены (рисунок 2), нос своеобразной крючковидной формы («клюв попугая»), гипоплазия верхней челюсти, псевдопрогения; в резко выраженных случаях наблюдается снижение умственного развития. Наследуется по доминантному типу.
Рентгенологически выявляются изменения черепа. На первый план выступает характерная деконфигурация головы и нарушение нормальных соотношений между мозговым и лицевым черепом: первый уменьшен в размерах, имеет почти шаровидную форму, швы заращены, усилены пальцевые вдавления. Кости свода черепа истончены, несколько выпячиваются кнаружи в области переднего родничка. Основание черепа укорочено и углублено, область турецкого седла сужена, глазницы уплощены.
 |
|  |
Рис. 2. | ||
Кости лицевого черепа малы: верхняя челюсть и носовые кости недоразвиты, нижняя челюсть значительно выдаётся вперёд, в силу чего образуется резкий прогиб носа внутрь.
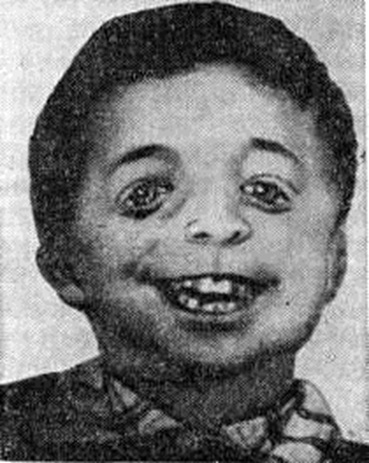
Челюстно-лицевой дизостоз (синдром Берри — Франческетти, синдром Франческетти — Цвалена) — гипоплазия главным образом нижней челюсти и скуловых костей, макростомия (своеобразное «рыбье» или «птичье» лицо), широкие косо расположенные глазные щели (рисунок 3), с вывороченными и скошенными книзу веками и колобомами в наружных отделах, слепые фистулы от углов рта к ушам, языковидное оволосение щёк, нарушения развития зубов, деформация ушных раковин, иногда среднего и внутреннего уха с развитием глухоты, устранимой операцией. В противоположность синдромам Крузона и Апера (смотри полный свод знаний Апера синдром) определяется сильное развитие лобных пазух. Встречается деформация грудной клетки и позвоночника. Наследуется по доминантному типу.
|
| |
Рис. 3. | ||
Челюстно-черепной дизостоз (синдром Петерс — Хевельса) — гипоплазия верхней челюсти, скуловых дуг, открытый прикус, прогения (выстояние нижней челюсти), укорочение переднего отдела основания черепа. Аномалия наследуется по доминантному типу.
Существуют другие формы черепных Дизостоз: синдромы Гегенхара, Робена, Франсуа и другие Внешний вид больных с различными формами Дизостоз характерен. Дизостоз сохраняется всю жизнь, не поддаётся оперативной коррекции, почти не требует дифференциальной диагностики с другими заболеваниями. В сомнительных случаях важным диагностическим методом является рентгено л. исследование.
Различают так называемый неполные типы перечисленных Дизостоз, когда имеют место не все характеризующие их симптомы. Отдельные признаки могут комбинироваться в различных сочетаниях, составляя как бы промежуточные типы Дизостоз
Прогноз для жизни благоприятный.
|
Виноградова Т.П.; Лагунова И.Г. |

|
⇐ Перейти на главную страницу сайта |
⇑ Вернуться в начало страницы ⇑ |
Библиотека Ordo Deus ⇒ |
⇐ Дизонтогенетические опухоли |
⇓ Полный свод знаний. Том первый А. ⇓ |
Дизурия ⇒ |
|
Все статьи в полном изложении, Вы можете найти в большой медицинской энциклопедии — Главный редактор: академик АН СССР (РАН) и АМН СССР (РАМН) Б.В. Петровский. — Москва издательство «Советская энциклопедия» 1989г. |
|
Внимание! Вы находитесь в библиотеке «Ordo Deus». Все книги в электронном варианте, содержащиеся в библиотеке «Ordo Deus», принадлежат их законным владельцам (авторам, переводчикам, издательствам). Все книги и статьи взяты из открытых источников и размещаются здесь только для чтения. |
|
Библиотека «Ordo Deus» не преследует никакой коммерческой выгоды. |
|
Все авторские права сохраняются за правообладателями. Если Вы являетесь автором данного документа и хотите дополнить его или изменить, уточнить реквизиты автора, опубликовать другие документы или возможно вы не желаете, чтобы какой-то из ваших материалов находился в библиотеке, пожалуйста, свяжитесь с нами по e-mail: |
Вас категорически не устраивает перспектива безвозвратно исчезнуть из этого мира? Вы не желаете закончить свой жизненный путь в виде омерзительной гниющей органической массы пожираемой копошащимися в ней могильными червями? Вы желаете вернувшись в молодость прожить ещё одну жизнь? Начать всё заново? Исправить совершённые ошибки? Осуществить несбывшиеся мечты? Перейдите по ссылке: «главная страница».
|
© Ordo Deus, 2010. При копировании ссылка на сайт http://www.ordodeus.ru обязательна. |