Миопатия |
||
 |
 |
|
Оглавление
|
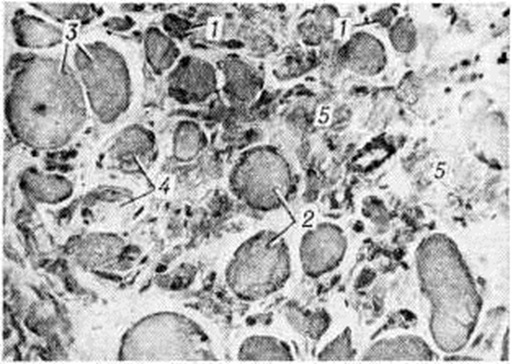
МиопатияМиопатия (греческий mys, my[os] мышца + -pathos страдание, болезнь) — группа наследственных заболеваний мышц, основными клиническими проявлениями которых являются мышечная слабость, атрофия, снижение мышечного тонуса, снижение или отсутствие сухожильных рефлексов, изменение биоэлектрической активности мышц. Первые клинико-морфологически описания заболеваний этой группы принадлежат Г. Дюшенну, Ж. Крювелье (1853), В. Эрбу (1883—1884), В. К. Роту (1876, 1895). Патология встречается во всех странах мира. Частота различных форм составляет 2—6 на 100 000 населения. Классификация Вопросы классификации Миопатия разрабатываются в различных направлениях. Миопатия классифицируют по типу наследования: аутосомно-рецессивные, аутосомно-доминантные, рецессивные и доминантные, сцепленные с Х-хромосомой. В зависимости от времени появления первых симптомов и характера течения Миопатия под разделяют на врождённую непрогрессирующую Миопатия и прогрессирующую мышечную дистрофию (ранняя детская, детская, юношеская и поздняя формы). Прогрессирующая мышечная дистрофия также подразделяется на формы в зависимости от преимущественной локализации миодистрофического процесса (например, плече-лопаточно-лицевая миодистрофия Ландузи — Дежерина, тазоплечевая миопатия Эрба — Рота, бульбарно-офтальмоплегическая Миопатия, дистальная Миопатия), характера распространения процесса восходящий и нисходящий варианты). В отдельную группу выделяют Миопатия с выраженной псевдогинертрофией мышц (псевдогипертрофическая миопатия Дюшенна, доброкачественная псевдогипертрофическая миопатия Беккера и другие). Этиология и патогенез Причиной заболеваний являются генетически детерминированные дефекты метаболизма мышечной ткани или структуры мышечных клеток. Поскольку первичный молекулярный дефект не известен ни для одной из форм Миопатия, патогенез детально не изучен. По-видимому, он не однозначен для всех форм Миопатия Известно, однако, что при всех заболеваниях этой группы в большей или меньшей степени выражены усиленный распад мышечных белков, преобладающий над их ускоренным, но неполноценным синтезом, изменения сосудов, нарушения проницаемости их стенок, нарушения структуры и проницаемости клеточных мембран, сдвиг обмена катионов, изменения в соединительной ткани и другие Предполагают, что при некоторых формах первичный молекулярный дефект локализуется не в самой мышечной ткани (а, например, в нервной) и служит лишь пусковым фактором указанных изменений. Возможно также, что при некоторых формах Миопатия эффект мутантного гена более генерализован и распространяется на мышечную, нервную и другие ткани. Патологическая анатомия Морфологически изменения при Миопатия характеризуются нарастающей атрофией скелетных мышц, которые уменьшаются в объёме и становятся плотными, бурого цвета вследствие разрастания соединительной ткани или, напротив, увеличиваются в объёме за счёт жировой клетчатки. При различных формах мышечных дистрофий (псевдогипертрофических, плече-лопаточно-лицевой, тазоплечевой, офтальмоплегической, бульбарно-офтальмоплегической) определяются в основном однотипные гисто л. изменения (рисунок 1): уменьшение количества мышечных волокон в пучках, резкая диффузная разнокалиберность сохранившихся волокон с преобладанием среди них атрофированных, гиалиновая и вакуольная дистрофия в части мышечных волокон, дискоидный и коагуляционный некроз отдельных волокон, расщепление гипертрофированных волокон, разрастание соединительной и жировой ткани в эндо и перимизии. В некоторых мышечных волокнах находят саркоплазматические тельца, саркоплазматические массы, кольцевидные миофибриллы. Изредка встречаются регенерирующие волокна с круглыми сочными ядрами и богатой рибонуклеопротеидами саркоплазмой. Мышечные веретена длительное время остаются неизмененными. В части наблюдений выявляются периваскулярные лимфоидно-гистиоцитарные инфильтраты. По мере нарастания двигательных нарушений отмечается постепенное уменьшение количества мышечных волокон и нивелировка их диаметра за счёт резкого уменьшения количества и калибра гипертрофированных волокон. |
Наиболее быстро эти изменения развиваются при миопатии Дюшенна — злокачественном варианте псевдогипертрофической Миопатия. Для доброкачественного варианта — миопатии Беккера — характерно сочетание выраженных процессов липоматоза и склероза с резкой гипертрофией части мышечных волокон.
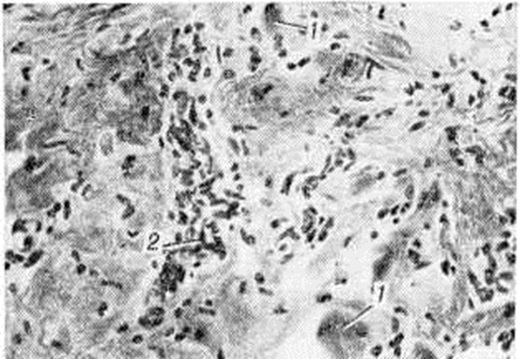
С последней особенностью связывают длительную компенсацию двигательных нарушений у таких больных. В далеко зашедшей стадии Миопатия определяются небольшие островки из атрофированных мышечных волокон на фоне резкого склероза и липоматоза эндои перимизия (рисунок 2), при этом гистологически не представляется возможным дифференцировать Миопатия с нейромышечной атрофией. У пробандов при биопсии мышц находят единичные атрофированные мышечные волокна преимущественно I типа, пролиферацию ядер с переходом их в центр волокна и незначительное увеличение соединительной ткани в эндомизии.
 |
|  |
Рис. 1. | ||
|
| |
Рис. 2. | ||
Наиболее ранние ультраструктурные изменения в мышцах при Миопатия характеризуются утолщением и расщеплением Z-линий с последующим разрушением миофибрилл мышечного волокна. Благодаря фазово-контрастной и электронной микроскопии выявлены дефекты в мембранных системах мышечного волокна. Гистоферментохимически отмечается ослабление реципрокных отношений между гликолитическими и окислительными ферментами в мышечных волокнах различного типа.
В центральной и периферической нервной системе изменений не обнаружено. Характерен кардиосклероз.
Клиническая картина
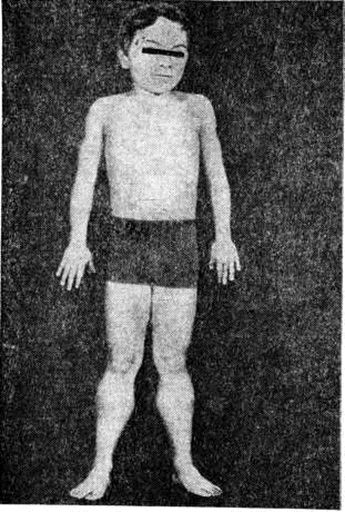
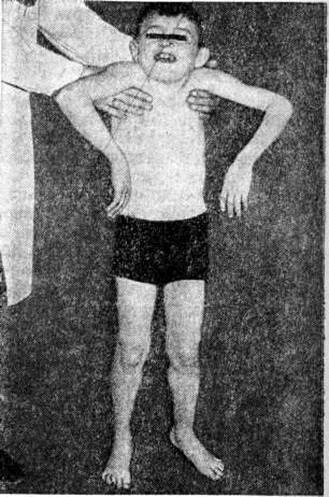
Ведущими симптомами заболеваний этой группы являются повышенная утомляемость и слабость мышц, симметричные мышечные атрофии, снижение или отсутствие сухожильных рефлексов. При отдельных формах заболевания отмечается псевдогипертрофия (рисунок 3), когда объем поражённых мышц увеличен, хотя сила их снижена так же, как при атрофии. При локализации миодистрофического процесса в области лица мимика больных становится бедной. Гипомимия приводит к характерному выражению лица — «миопатическое лицо». Следствием атрофии круговой мышцы рта является «поперечная улыбка». Губы утолщены и несколько вывернуты кнаружи — «губы тапира». На лбу отсутствуют морщины — симптом «полированного лба».
|
| |
Рис. 3. | ||
|
| |
Рис. 4. | ||
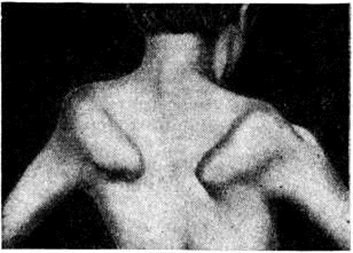
Поражение поперечнополосатых мышц глаз приводит к частичной или полной офтальмоплегии, птозу, экзофтальму, лагофтальму. Поражение мышц мягкого неба, глотки и гортани проявляется нарушением глотания и фонации. Симптомы поражения мышц плечевого пояса — ограничение объёма активных движений в проксимальных отделах рук, отставание лопаток от туловища — симптом «крыловидных лопаток» (рисунок 4), отсутствие сопротивления мышц плечевого пояса при поднимании больного за подмышки — симптом «свободных надплечий» (рисунок 5.); плечи больного поднимаются вверх, а голова как бы проваливается между ними. Атрофия длинных мышц спины и тазового пояса проявляется нарушением осанки и походки: выражен гипер лордоз позвоночника, голова несколько запрокинута назад, туловище при ходьбе ритмично раскачивается — «утиная походка». Затруднено поднимание по лестнице, вставание из сидячего положения. Для того чтобы принять вертикальное положение, больной вынужден прибегать к помощи рук, опираясь на соседние предметы или собственные бедра,— вставание «лесенкой» (симптом «лестницы» — рисунок 6, я, б, в). При атрофии косых мышц живота наблюдается симптом «осиной талии». Нарушение походки по типу «степпажа» или «петушиной походки» характерно для локализации миодистрофического процесса в мышцах голени и стопы. Поражение мышц приводит к ограничению подвижности суставов вплоть до образования контрактур. Присоединяющаяся, как правило, в поздней стадии заболевания легочно-сердечная недостаточность является следствием миодистрофического процесса в миокарде и дыхательной мускулатуре. При электромиографическом исследовании выявляют снижение амплитуды осцилляций, высокую частоту полифазных потенциалов, укорочение времени отдельных осцилляций.
Клинико-морфологическая характеристика некоторых наиболее распространённых форм. Наряду с общими клинические, и морфологический признаками, характерными для миопатий, отдельные формы имеют отличительные клинико-морфологические признаки; установление формы непрогрессирующей врождённой Миопатия возможно лишь на основании детального гистологический исследования мышц.
|
| |
Рис. 5. | ||
|

| |
Рис. 6. | ||
Миопатия Эрба Рота — прогрессирующая мышечная дистрофия, проксимальная, аутосомно-рецессивная. Гистологической особенностью её является образование гигантских мышечных волокон с последующим их расщеплением, в результате которого образуются группы мелких волокон. Мальчики болеют несколько чаще девочек. В зависимости от времени появления первых симптомов выделяют раннюю детскую, детскую и юношескую формы. Характерно поражение гладкой мускулатуры кишечника, развитие сердечно-лёгочной недостаточности, контрактур крупных суставов при относительно длительной сохранности мышц дистальных отделов конечностей и лица. Форма заболевания, начинающаяся с поражения мышц тазового пояса с восходящим типом поражения, известна в литературе под названием миопатия Лейдена — Мебиуса (смотри полный свод знаний Лейдена дистрофия).
Миопатия Дюшенна — прогрессирующая мышечная дистрофия, ранняя детская, проксимальная, псевдогипертрофическая, рецессивная, сцепленная с Х-хромосомой. Болеют мальчики. Аналогичное заболевание, встречающееся у девочек, считают псевдогипертрофической аутосомно-рецессивной формой. Характерной особенностью патоморфологические изменений в мышце уже на ранних стадиях процесса является разрастание жировой и соединительной ткани и замещение ими мышечной, чем и объясняется наличие псевдогипертрофии. Заболевание характеризуется появлением слабости и атрофии мышц в возрасте до 3 лет, злокачественным течением и наличием псевдогипертрофий, особенно в икроножных мышцах. У больных могут быть нейроэндокринные нарушения в виде ожирения, гипергидроза и другие
Частый симптом заболевания — отставание в умственном развитии. Рано поражается миокард, может быть дисфункция диафрагмы и гладких мышц желудочно-кишечного тракта и мочевого пузыря. В плазме крови и моче значительно повышена активность мышечных ферментов.
Миопатия Беккера — прогрессирующая мышечная дистрофия, поздняя, доброкачественная, проксимальная, псевдогипертрофическая, рецессивная, сцепленная с Х-хромосомой. Заболевание проявляется в возрасте 20—30 лет, прогрессирует медленно; эндокринные нарушения, поражение миокарда и снижение интеллекта не характерны.
Миопатия Ландузи — Дежерина — прогрессирующая мышечная дистрофия плече-лопаточно-лицевая, ювенильная, медленно прогрессирующая, аутосомно-доминантная. Гистологически в отличие от других прогрессирующих мышечных дистрофий обычно находят увеличение среднего диаметра волокон и появление мелких волокон, характерных для денервации.
Первые симптомы заболевания появляются обычно в возрасте 12—20 лет, характерна длительная сохранность двигательных функций. В зависимости от преимущественного поражения тех или иных групп мышц и последовательности распространения патологический процесса различают следующие формы заболевания: лице-лопаточно-плечевую, лице-лопаточно-плече-перонеальную, лице-лопаточно-плече-ягодично-бедренную, лице-лопаточно-плече-ягодично-бедренно-перонеальную и лице-лопаточно-плече-перонеально-ягодично-бедренную. Вариант заболевания с более поздним началом (27—30 лет), при котором первоначально поражаются малоберцовые мышцы, а затем, в меньшей степени, мышцы плечевого пояса и лица, называют лопаточно-перонеальной формой миопатии Давиденкова.
Миопатия Говерса — Веландера (миопатия Невина) — прогрессирующая мышечная дистрофия, дистальная, поздняя, аутосомно-доминантная. Ранние гистологический изменения напоминают изменения при миотонической дистрофии (смотри полный свод знаний Миотония), но по мере прогрессирования заболевания преобладающим становится миодистрофический процесс. Симптомы поражения мышц дистальных отделов конечностей появляются в возрасте 30—60 лет, причём почти исключительно поражаются разгибатели; распространение патологический процесса, как правило, нисходящее, но если сначала поражаются мышцы ног, то быстро прогрессирует нарушение походки. Миокард поражается не постоянно.
Миопатия Кило — Невина — прогрессирующая мышечная дистрофия, офтальмоплегическая, ювенильная, аутосомно-доминантная. Отличительной гистологический особенностью офтальмоплегической Миопатия, в частности бульбарно-офтальмоплегической формы, является присутствие в мышечных волокнах большого количества так называемый окаймлённых вакуолей, которые выглядят как отверстия от компостера, окружённые материалом, окрашивающимся трихромовым красителем в ярко-красный цвет. Первые симптомы заболевания в виде двустороннего птоза появляются в возрасте до 20 лет. По мере прогрессирования заболевания поражаются мышцы глазного яблока, что приводит к полной наружной офтальмоплегии. В 25% случаев процесс распространяется на круговую мышцу глаза, другие лицевые мышцы, мышцы глотки и гортани (бульбарно-офтальмоплегическая форма Миопатия) и мышцы плечевого пояса. В поздней стадии заболевания могут поражаться также мышцы плечевого и тазового пояса. Описаны также случаи заболевания с более поздним началом и более доброкачественным течением.
|
| |
Рис. 7. | ||
|
| |
Рис. 8. | ||
Миопатия непрогрессирующая врождённая— группа заболеваний, характеризующихся врождёнными нарушениями структуры или метаболизма мышечной ткани, диффузным поражением мышц, отсутствием прогрессирования или очень медленно прогрессирующим течением, в редких случаях с возрастом симптомы заболевания могут становиться менее выраженными.
Среди заболеваний этой группы на основе гистологический данных выделяют несколько форм.
Болезнь центрального стержня, гистологически характеризующаяся присутствием в мышечных волокнах центральных участков, которые окрашиваются эозином интенсивнее, чем остальная саркоплазма, лишены гликогена и ферментативной активности. Электронно-микроскопически стержень представлен аморфными и неправильно расположенными фрагментами миофиламентов и Z-линий, митохондрии и саркоплазматический ретикулум отсутствуют.
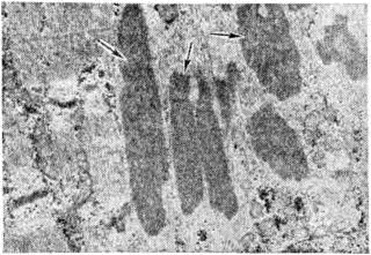
Немалиновая Миопатия, характеризующаяся наличием в субсарколеммных зонах части мышечных волокон нитевидных структур (рисунок 7). Гистохимически в них выявлены рибонуклеопротеиды и высокая активность тирозиназы.
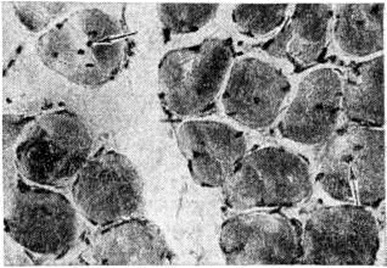
Миотубулярная (центронуклеарная) Миопатия, характеризующаяся тем, что 30—85% мышечных волокон содержат центрально расположенные ядра, вокруг которых отсутствуют миофибриллы (рисунок 8); в целом такие волокна напоминают миотубулы мышц плода, что послужило основанием для предположения о том, что в основе заболевания лежит отсутствие созревания и дифференцировки мышечной ткани.
Врождённая диспропорция типов волокон, характеризующаяся небольшим уменьшением диаметра волокон I гистологический типа и увеличением диаметра волокон II гистологический типа.
5. Митохондриальные миопатии — группа врождённых медленно прогрессирующих заболеваний, при которых основные изменения в мышечной ткани связаны с аномалиями митохондрий. Среди заболеваний этой группы выделяют: 1) плеокониальную Миопатия, характеризующуюся тем, что митохондрии мышечных волокон содержат множественные кристы и иногда круглые осмиофильные включения; 2) мегакониальную Миопатия, характеризующуюся присутствием в мышечных волокнах скоплений увеличенных причудливой формы митохондрий, которые могут содержать необычные включения; 3) митохондриальную Миопатия с гиперметаболизмом, характеризующуюся присутствием в перинуклеарных участках мышечной клетки гигантских митохондрий, которые могут быть трёх типов: 1-й тип — с тубулярным типом крист, 2-й тип — с непрозрачными тельцами, 3-й тип— состоящие из плотных концентрических тяжей. Гистохимически в мышцах выявляется высокая активность окислительно восстановительных ферментов.
Диагноз
Диагноз ставится на основании клинической картины и данных гистологического исследования мышечной ткани (при непрогрессирующей врождённой Миопатия диагноз в основном базируется на данных гистологии биоптата мышц). Заболевания из группы Миопатия следует дифференцировать со спинальными и невральными амиотрофиями (смотри полный свод знаний), миозитами (смотри полный свод знаний), дерматомиозитом (смотри полный свод знаний), врождёнными атрофиями мышц (смотри полный свод знаний Атрофия мышечная), последствиями полиомиелита (смотри полный свод знаний), врождённым миосклерозом Ловенталя, который характеризуется врождённой симметричной тугоподвижностью суставов вследствие склерозирования мышц и кожи. От Миопатия следует отличать также клинически сходные заболевания из группы гликогенозов и Миопатия, обусловленные нарушением обмена липидов с известными первичными молекулярными дефектами. Среди первых наиболее распространённой и изученной является болезнь Мак-Ардла (смотри полный свод знаний Гликогенозы). К миопатиям, обусловленным накоплениями липидов, относятся наследственный дефицит карнитина, наследственный дефицит карнитин-пальмитоил-трансферазы, наследственный дефицит пируватдекарбоксилазы, болезнь накопления нейтральных липидов. Заболевания этой группы проявляются мышечной слабостью и (или) миоглобинурией, атрофией мышц, анорексией, гипотрофией, возможны увеличение печени и признаки сердечной и почечной недостаточности. Патоморфологически при них выявляются дистрофические изменения в поперечнополосатых мышцах, в миокарде и отложение в них липидов, некроз и дистрофия почечных канальцев, накопление в них жиров, жировая инфильтрация печени. Для диагностики этих заболеваний необходимо исследование активности ферментов. Материалом для исследования могут служить кожные фибробласты, мышцы, лимфоциты крови.
Лечение
Лечение всех форм Миопатия является патогенетическим и должно быть направлено на улучшение трофики мышечной ткани и проведения импульса по нервному волокну и через мионевральный синапс. Для улучшения трофики мышц назначают комплексы аминокислот (глутаминовую кислоту, лейцин, метионин), АТФ, анаболические стероиды, оротат калия, антихолинэстеразные препараты (прозерин, галантамин, оксазил, нива лип, калимин), витамины группы В. Рекомендуются повторные переливания донорской крови, электростимуляция мышц, прозерин-электрофорез. С целью улучшения местного кровообращения применяют оксигенобаротерапию, ультразвук, массаж, тепловые процедуры, бальнеолечение. Лечение проводят постоянно курсами, препараты назначают с учётом возраста больного, формы заболевания, степени тяжести клинические, проявлений Миопатия
Прогноз
Прогноз определяется быстротой прогрессирования дистрофических изменений в скелетной мускулатуре, а также возрастом, в котором начинается заболевание. При наиболее злокачественной форме миопатии Дюшенна уже в детском возрасте развивается полная обездвиженность больных. Летальный исход может быть обусловлен нарастанием лёгочной и сердечной недостаточности, гиповентиляционной и гипостатической пневмонией. При поздних формах, начинающихся после 20— 30 лет, течение относительно доброкачественное, больные могут длительное время сохранять трудоспособность. Миотубулярная, немалиновая Миопатия, «болезнь центрального стержня», митохондриальные Миопатия обычно отличаются медленным прогрессированием, с возрастом могут приобретать стационарный характер.
|
Бадалян Л.О.; Копьева Т.H. |

|
⇐ Перейти на главную страницу сайта |
⇑ Вернуться в начало страницы ⇑ |
Библиотека Ordo Deus ⇒ |
⇐ Миома |
⇓ Полный свод знаний. Том первый А. ⇓ |
Миоренальный синдром ⇒ |
|
Все статьи в полном изложении, Вы можете найти в большой медицинской энциклопедии — Главный редактор: академик АН СССР (РАН) и АМН СССР (РАМН) Б.В. Петровский. — Москва издательство «Советская энциклопедия» 1989г. |
|
Внимание! Вы находитесь в библиотеке «Ordo Deus». Все книги в электронном варианте, содержащиеся в библиотеке «Ordo Deus», принадлежат их законным владельцам (авторам, переводчикам, издательствам). Все книги и статьи взяты из открытых источников и размещаются здесь только для чтения. |
|
Вся информация на сайте Ordo Deus находится в свободном доступе. Ordo Deus не предоставляет информацию на платной основе. |
|
Все авторские права сохраняются за правообладателями. Если Вы являетесь автором данного документа и хотите дополнить его или изменить, уточнить реквизиты автора, опубликовать другие документы или возможно вы не желаете, чтобы какой-то из ваших материалов находился в библиотеке, пожалуйста, свяжитесь с нами по e-mail: |
Вас категорически не устраивает перспектива безвозвратно исчезнуть из этого мира? Вы не желаете закончить свой жизненный путь в виде омерзительной гниющей органической массы пожираемой копошащимися в ней могильными червями? Вы желаете вернувшись в молодость прожить ещё одну жизнь? Начать всё заново? Исправить совершённые ошибки? Осуществить несбывшиеся мечты? Перейдите по ссылке: «главная страница».
|
© Ordo Deus, 2010. При копировании ссылка на сайт http://www.ordodeus.ru обязательна. |