Нефротический синдром |
||
 |
 |
|
Оглавление
|
Нефротический синдромНефротический синдром (греческий nephros почка; синдром) — неспецифический симптомокомплекс, характеризующийся массивной протеинурией (5 и более граммов в сутки) и нарушениями белково-липидного и водно-солевого обмена, проявляющимися гипоальбуминемией, диспротеинемией, гиперлипидемией, липидурией, отёками и водянкой серозных полостей. История вопроса о данном симптомокомплексе насчитывает более 70 лет, однако термин «нефротический синдром» фигурирует в литературе лишь с 1949 год Термин получил мировое признание, почти полностью заменив старый термин «нефроз», и в 1968 год был введён в номенклатуру болезней ВОЗ. Однако и старый термин «нефроз» ещё не полностью утратил своё значение. Его употребляют патологоанатомы, особенно применительно к амилоидозу почек, и педиатры, многие из которых пользуются термином «липоидный нефроз». Последний согласно современной классификации болезней почек применяется для обозначения первичного Нефротический синдром у детей и взрослых, развивающегося на основе минимальных клубочковых изменений. Учение о липоидном нефрозе как о дистрофическом изменении канальцевого эпителия нашло продолжение в том, что многие формы поражений почек, развивающихся в связи с токсическими и некротическими их повреждениями (нефроз токсический, нефроз миоглобинурийный, нефроз парапротеинемический, нефроз сифилитический и другие) стали трактоваться так же. В Международной классификации болезней их относят к группе нефропатий с уточнением их этиологии или к группе острой почечной недостаточности с указанием на наличие некротических изменений той или иной локализации. Чаще Нефротический синдром болеют дети в возрасте от 2 до 5 лет и взрослые от 17 до 35 лет. Оглавление Врождённый (семейный) нефротический синдром Экспериментальный нефротический синдром Этиология и патогенезНефротический синдром подразделяют на первичный и вторичный. Первичный Нефротический синдром развивается при таких заболеваниях почек, как гломерулонефрит, липоидный нефроз, мембранозная нефропатия, IgA-нефропатия, врождённый, семейный нефротический синдром, нефропатическая форма первичного амилоидоза. Вторичный Нефротический синдром обусловлен многочисленными заболеваниями. К ним относятся системная красная волчанка, узелковый периартериит, системная склеродермия, ревматизм, ревматоидный артрит, геморрагический васкулит, затяжной септический эндокардит, хронический воспалительные заболевания, туберкулёз, сифилис, гепатит и другие Более редкой причиной Нефротический синдром являются лимфогранулематоз, миеломная болезнь, тромбоз вен и артерий почек, аорты или нижней полой вены; опухоли различной локализации; аллергические заболевания. Вторичный Нефротический синдром может развиваться при нефропатии беременных, а также при сахарном диабете (на почве диабетического гломерулосклероза). Дискутируется вопрос о возможностях и механизмах развития Нефротический синдром при пиелонефрите. Определённую проблему составляет развитие гломерулонефрита в почечном аллотрансплантате, нередко в связи с Нефротический синдром |
Морфологически основой при вторичном Нефротический синдром может быть специфическая нефропатия (волчаночная, ревматоидная и другие), амилоидоз почек, гломерулонефрит или сочетанная патология (как при узелковом периартериите). Поэтому гистологический картина при световой, иммунофлюоресцентной и электронной микроскопии отражает признаки не только самого Нефротический синдром, но и изменения, свойственные названным заболеваниям.
Большинство заболеваний, обусловливающих Нефротический синдром, возникает на иммунной основе, то есть вследствие осаждения в органах (в том числе в почках) иммунных комплексов или вследствие взаимодействия антител с антигенами базальной мембраны капилляров клубочков с сопутствующими нарушениями клеточного иммунитета. Последние проявляются в реакциях органоспецифической гиперчувствительности замедленного типа и в ослаблении контролирующего пула Т-лимфоцитов. Иммунные механизмы активируют гуморальные и клеточные звенья воспалительной реакции и сосудистой проницаемости с развитием клеточной эмиграции, фагоцитоза, дегрануляции лейкоцитов и освобождением при этом лизосомальных ферментов, способных повреждать структуры тканей (смотри полный свод знаний Воспаление). Антигены, входящие в состав иммунных комплексов, могут быть экзо и эндогенными. К экзогенным относят антигены бактериальные, вирусные, паразитарные, медикаментозные, пищевые, пыльцу растений, соединения тяжёлых металлов и другие Эндогенными антигенами при ряде заболеваний могут стать тиреоглобулин, ДНК, денатурированные нуклеопротеиды, криоглобулины, белки опухолевого происхождения.
Антитела к этим антигенам в большинстве случаев принадлежат к классу IgM или одновременно к нескольким классам Ig.
Величина иммунных комплексов зависит от характера антигена и связанных с ним антител. Малые комплексы обыкновенно содержат избыток антигенов и растворимы. Большие комплексы, мол. вес которых больше 50 000, содержат избыток антител, легко депонируются в стенках микрососудов органов, в том числе почек, вызывая развитие вторичных воспалительных реакций (нефропатий). Степень поражений органа зависит от концентрации комплексов, их состава и продолжительности антигенной стимуляции.
Однако не все заболевания, вызывающие Нефротический синдром, имеют доказанный иммунокомплексный генез. Так, не ясен патогенез липоидного нефроза, врождённого Нефротический синдром финского типа, Нефротический синдром при таких генетически обусловленных болезнях, как мукополисахаридозы или парциальная липодистрофия.
Изучаются иммуногенетические аспекты патогенеза Нефротический синдром различного происхождения. Типирование по системе HLA больных с Нефротический синдром показало существенное преобладание определённых антигенов системы гистосовместимости при ряде нозологических форм нефротического синдрома: при нефротическом синдроме на почве геморрагического васкулита преобладали HLA—BW35, среди больных с атопическим Нефротический синдром у большей половины выявляли HLA—В12, при системной красной волчанке — HLA—38. Однако, по данным Томсона (P. D. Thomson) с соавторами (1976) и Шерака (Одонтома Scherak) с соавторами (1978), корреляции между клиническими, иммунологическими показателями и теми или иными антигенами системы HLA выявлено не было.
Если иммунологический концепция патогенеза применима для большинства нозологических форм, течение которых осложняет Нефротический синдром, то механизмы большой нефротической протеинурии нельзя признать окончательно выясненными. Определёнными вехами в учении о патогенезе Нефротический синдром являются: концепция обменно-дискразическая; концепция эндокринной недостаточности; иммунологическая (в большей мере применимая к обусловливающей Нефротический синдром нефропатии); метаболическая, или физико-химическая, являющаяся наиболее признанной.
Отправным пунктом метаболической концепции патогенеза является общепризнанный факт, что нефротическая протеинурия обусловлена главным образом усиленной проницаемостью клубочкового фильтра. Усиление клубочковой проницаемости при Нефротический синдром, как установлено, прежде всего связано с уменьшением постоянного электрического заряда стенки капиллярной петли. Последнее обусловлено исчезновением из неё сиалопротеина, в норме тонким слоем покрывающего эндотелий и его отростки, лежащие на базальной мембране, и входящего также в состав самой мембраны.
Исследование химический состава базальных мембран при различных формах нефротического синдрома позволило установить увеличение содержания коллагена в базальной мембране и активности ферментов, принимающих участие в его синтезе, а также уменьшение содержания в ней 3-гидроксипролина, 4-гидроксипролина, глицина.
Предполагается, что на местах максимальной потери анионов скапливаются полиморфно-ядерные лейкоциты, лизосомальные ферменты которых разрушают материал базальной мембраны, вследствие чего в мочу поступают фрагменты клубочковой базальной мембраны. Изменённые, распластанные по базальной мембране подоциты (размеры их могут быть в 7—15 раз больше по сравнению с нормой) не полностью закрывают места разрушения, через которые и происходит утечка высокомолекулярного белка. Синтез вещества базальной мембраны подоцитами и (или) мезангиальными клетками снижен и извращён. При большой фильтрации белков через мембраны капилляров клубочков проксимальные канальцы не в состоянии реабсорбировать и деградировать белок, что ведёт к развитию тяжёлой гиалиново-капельной и вакуольной дистрофии эпителия.
Патологическая анатомия
При нефротическом синдроме первичными являются изменения гломерулярного фильтра, с ними связана нарастающая протеинурия.
Изменения канальцев, стромы, сосудов вторичны и развиваются в связи с реабсорбционной тубулоинтерстициальной недостаточностью и со все возрастающей в этих условиях гипоксией почечной ткани. Изменения почек при Нефротический синдром, рассматриваемые как протеинурические повреждения, хорошо прослеживаются в динамике на ультраструктурном и клеточном уровнях.
Протеинурия, обусловленная избыточной фильтрацией белков плазмы, превосходящей реабсорбционные возможности канальцевого эпителия, вызывает структурную перестройку гломерулярного фильтра и канальцевого аппарата.
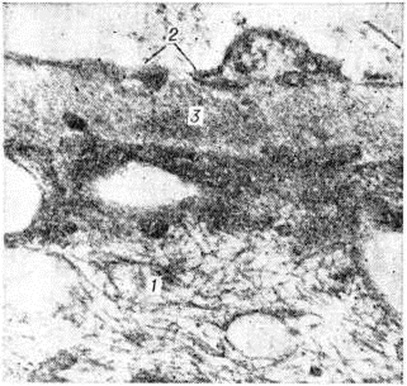
При протеинурии в цитоплазме подоцитов появляется множество пиноцитозных пузырьков, выявляется хорошо развитая цитоплазматическая сеть, обилие рибосом и полисом, усиливается фибриллярный рисунок цитоплазмы, причём фибриллы ориентированы по оси возможного сокращения клеток-насосов (рисунок 1). Эти ультраструктурные изменения свидетельствуют о повышенной функциональный активности подоцитов. Декомпенсация функции подоцитов ведёт к повреждению эндотелия, он вакуолизируется, набухает, происходит его десквамация, что сопровождается компенсаторной пролиферацией клеток эндотелия.
Повреждение гломерулярного фильтра сопровождается адаптивной гиперплазией мезангиальных клеток, продуцирующих мембраноподобное вещество мезангиального матрикса и вещество базальной мембраны. Отложение этого вещества в мезангии и очаговое утолщение базальной мембраны вблизи активных мезангиальных клеток дополняют структурную адаптивную перестройку гломерулярного фильтра при Нефротический синдром
 |
|  |
Рис. 1. | ||
|
| |
Рис. 2. | ||
|
| |
Рис. 3. | ||
|
| |
Рис. 4. | ||
|
| |
Рис. 5. | ||
|
| |
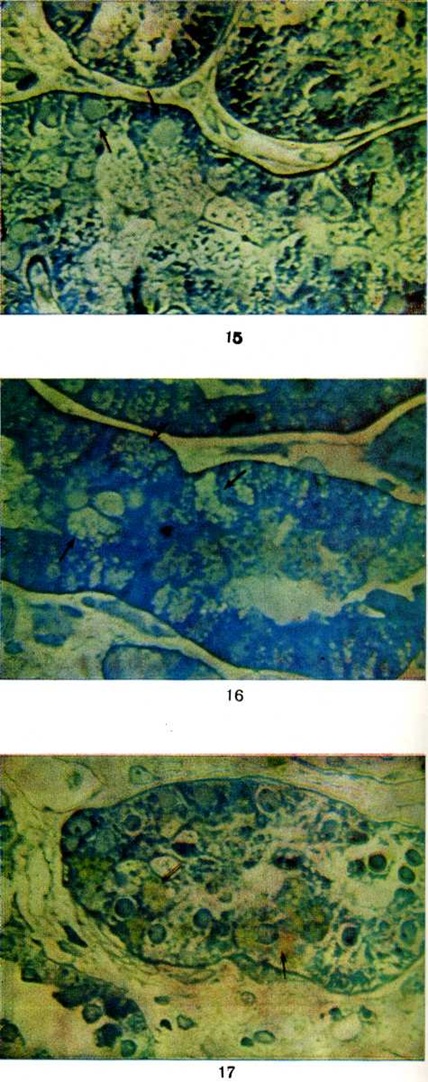
Рис. 15. Микропрепарат почки при нефротическом синдроме: гиалиновокапельная дистрофия (указано стрелками) эпителия канальцев главных отделов нефрона. Полутонкий срез, окраска метиленовым синим-азур II-фуксином; × 400. | ||
Морфологически эквивалентом протеинурии и истощения резорбтивной функции эпителия канальцев является гиалиново-капельная, вакуольная, баллонная и жировая дистрофия эпителия (цветной рисунок 1, 2, 3), при которых активность .ферментов в эпителии нефронов резко снижена (смотри полный свод знаний Дистрофия клеток и тканей). Электронно-микроскопически обнаруживаются набухание, вакуолизация и распад митохондрий, разрыв цистерн цитоплазматической сети, разрушение мембран. В результате дистрофических процессов развивается некробиоз и десквамация эпителия, которые являются основой формирования цилиндров, обтурирующих просветы канальцев, что приводит к кистозному их расширению и атрофии.
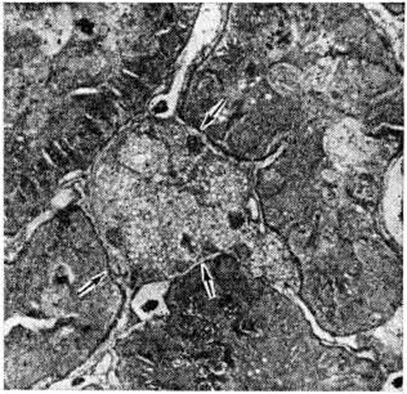
Отражением функциональный недостаточности лимфатических, системы почек — второй системы реабсорбции при Нефротический синдром служит отёк интерстиция, который быстро сменяется склерозом, причём среди разрастаний соединительной ткани часто встречаются большие светлые клетки с пенистой цитоплазмой (рисунок 2), которые считают макрофагами, фагоцитировавшими липиды. В сосудах почек находят плазматическое пропитывание и гиалиноз, склероз стенок.
Первичный нефротический синдром. Морфология первичного Нефротический синдром складывается из изменений, характерных для следующих его форм: липоидный нефроз, фокальный сегментарный гломерулярный гиалиноз, мембранозный гломерулонефрит (мембранозная нефропатия), врождённый Нефротический синдром (сведения о нем см. ниже).
Липоидный нефроз (синонимы: идиопатический Нефротический синдром детей, нефропатия с минимальными изменениями) описан впервые Мунком (F. Munk, 1913), который обнаружил в моче больных и в эпителии канальцев липиды. Он полагал, что изменения в почках связаны с общими нарушениями обмена.
Долгое время термины «липоидный нефроз», «мембранозный гломерулонефрит», «второй тип нефрита Эллиса», «нефротический синдром» употреблялись как синонимы. Благодаря работам Джонса (D. В. Jones, 1957) было выделено несколько форм Нефротический синдром: минимальные гломерулярные изменения, мембранозный гломерулонефрит и лобулярный гломерулонефрит.
Название «липоидный нефроз» было оставлено только для обозначения своеобразной патологии детей, проявляющейся Нефротический синдром с минимальными изменениями в клубочках почек, выявляемых при светооптическом исследовании. Термины «липоидный нефроз» и «минимальные изменения» стали использоваться как синонимы.
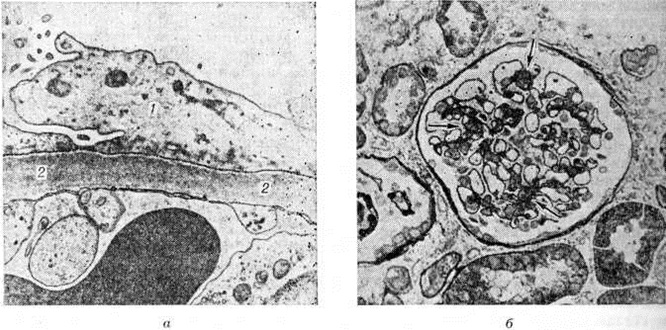
Сущность минимальных изменений установлена с помощью электронной микроскопии материала биопсии почек. При липоидном нефрозе изменяются только подоциты, у которых происходит слияние малых отростков, базальная мембрана при этом остаётся неизменённой (рисунок 3, а). После нескольких лет болезни к минимальным изменениям присоединяется очаговое утолщение базальных мембран капилляров (рисунок 3, б), увеличение мезангиального матрикса или количества мезангиальных клеток. Если заболевание ведёт к почечной недостаточности, в клубочках обнаруживают фокальный сегментарный склероз капилляров.
В эпителии проксимальных канальцев на ранних стадиях болезни выявляют двоякопреломляющие липиды и гранулы резорбированного белка. Со временем липиды исчезают из эпителия, появляются признаки атрофии канальцев, которая никогда не бывает значительной. Интерстиций ночек отёчен, к отеку присоединяется разрастание соединительной ткани, в которой находят пенистые клетки. При длительном течении болезни встречается утолщение внутренней оболочки сосудов.
Вид почек при липоидном нефрозе, протекающем без почечной недостаточности, характерен: они увеличены, очень бледные, поверхность их гладкая, на разрезе ткань набухшая, отёчная, жёлто-белая или бледно-серая — большая белая почка (смотри полный свод знаний Гломерулонефрит). В случаях смерти от почечной недостаточности почки немного уменьшены, плотные, поверхность их гладкая; ткань почек серого цвета, на разрезе выявляется жёлтая пятнистость.
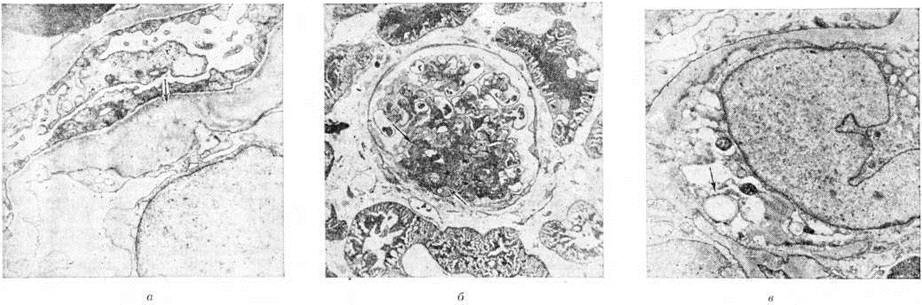
Фокальный сегментарный гломерулярный гиалиноз (очаговый склерозирующий гломерулонефрит) характеризуется преимущественным поражением юкстамедуллярных клубочков. Характерные для него изменения в виде сегментарного склероза впервые были описаны Ричем (A. R. Rich, 1957) у детей с липоидным нефрозом. Позднее Хабиб (R. Habib) с соавторами (1971) предложили для названия этих изменений термин «сегментарный гиалиноз». В процесс вовлекаются отдельные юкстамедуллярные клубочки (фокальные изменения), в которых склерозируются отдельные сегменты сосудистого пучка (сегментарные изменения); остальные клубочки интактны. В начале заболевания на светооптическом уровне изменения оцениваются как минимальные; электронно-микроскопически в материале биопсии почки находят характерные изменения базальной мембраны капилляров: неровные контуры эндотелиальной поверхности базальной мембраны (рисунок 4, а). При выраженной морфологический картине в отдельных капиллярах клубочков появляется гиалиновый материал в виде шаровидных отложений, обычно тесно связанных с капсулой клубочка (рисунок 4, б). В клубочках обнаруживаются пенистые клетки — мезангиальные клетки, содержащие липиды (рисунок 4, в) такие же клетки появляются и в интерстиции.
При иммуногистохимическом исследовании в капиллярах клубочков находят IgM, поэтому не исключают участия первичного иммунного механизма в развитии гломерулярных изменений.
По мере усиления интенсивности болезни в процесс вовлекаются клубочки поверхностных отделов коркового вещества. Сначала развивается склероз отдельных сосудистых петель, затем он охватывает все сосудистые петли клубочка (глобальный склероз). В канальцах находят жировую и белковую дистрофию эпителия, гиалиновые цилиндры в просветах, встречаются мелкие очаги кальцификации. Патогномонично образование очагов коллапса и атрофии канальцев, сопровождающееся склерозом стромы. Распространённость изменений канальцев пропорциональна выраженности изменений в клубочках.
Макроскопический вид почек тот же, что и при липоидном нефрозе.
Мембранозный гломерулонефрит характеризуется различными морфологический изменениями (смотри полный свод знаний Гломерулонефрит).
Вторичный нефротический синдром. Морфологической основой вторичного Нефротический синдром является гломерулонефрит, который может быть первичный или вторичный (при малярии, лейшманиозе, бактериальном эндокардите, ревматизме, системной красной волчанке, узелковом периартериите, геморрагическом васкулите, нефропатии беременных, гепатите, циррозе печени, тромбозе почечных вен, опухолях и так далее). По своему генезу в большинстве случаев это иммунокомплексный гломерулонефрит, обычно с подострым и хроническим, иногда острым течением. Гистологически при таком гломерулонефрите выявляются различные типы, однако преобладают экстракапиллярный продуктивный, мембранозный, мезангиокапиллярный и фибропластический; определённую специфику имеет волчаночный нефрит. Гломерулонефрит антительного генеза при Нефротический синдром встречается редко, прежде всего при синдроме Гудпасчера. В таких случаях при гистологический исследовании находят пролиферативный экстра или интракапиллярный типы гломерулонефрита. При Нефротический синдром, осложняющем гломерулонефрит любого генеза, резко выражены дистрофические изменения канальцев, слущивание эпителия, образование цилиндров. В тех случаях, когда резко выражена гидропическая дистрофия канальцевого эпителия, принято говорить о гидропическом нефрозе. Его описывали при туберкулёзе, эндокринопатиях, авитаминозах, голодании, но особенно часто при хронический поражениях кишечника, сопровождающихся диареей (нефроз кишечного истощения).
При хроническом пиелонефрите развитие Нефротический синдром связано не столько с тубулоинтерстициальными изменениями, сколько с инвазивным гломерулитом, ведущим к тяжёлым изменениям базальной мембраны и подоцитов гломерулярного фильтра.
Амилоидоз (смотри полный свод знаний), как и гломерулонефрит, одинаково часто является основным морфологический проявлением вторичного Нефротический синдром, причём именно нефропатический тип амилоидоза (амилоидоз почек, или амилоидный нефроз), независимо от того, является ли он первичным, генетическим, или вторичным.
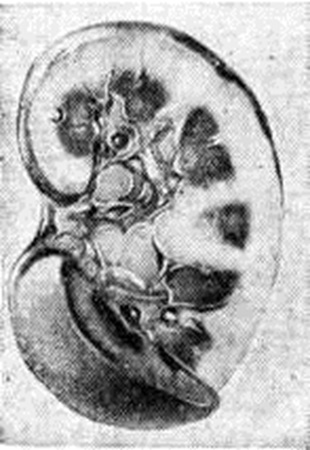
Развитие Нефротический синдром при амилоидозе связано с появлением амилоидного вещества в гломерулярном фильтре, при этохамилоидобластами, продуцирующими белок фибрилл амилоида, становятся мезангиальные клетки. Появлению амилоида в клубочках предшествует амилоидоз и склероз мозгового вещества и пограничного слоя почек, что ведёт к выключению и атрофии глубоко расположенных нефронов, редукции путей юкстамедуллярного кровотока и пирамидного лимфотока. Развивается гиалиново-капельная или вакуольная дистрофия эпителия канальцев: почки увеличиваются в размерах, становятся плотными; поверхность их бледно-серая или жёлто-серая. На разрезе корковое вещество широкое, матовое, мозговое вещество серо-розовое, сального вида (большая сальная почка — рисунок 5). При нарастании протеинурии и переходе протеинурической стадии амилоидоза почек в нефротическую стадию количество амилоида в почках увеличивается. Он обнаруживается во многих капиллярных петлях большинства клубочков, в артериолах и артериях, по ходу собственных мембран канальцев, но выраженные склеротические изменения коркового вещества отсутствуют. В пирамидах, наоборот, склероз и амилоидоз имеют диффузный характер. В эпителии канальцев наряду с гиалиново-капельной и вакуольной отмечается жировая дистрофия.
В эпителии канальцев и строме много двоякопреломляющих липидов (холестерина). Канальцы расширены, забиты цилиндрами. Почки становятся большими, плотными, восковидными (большая белая амилоидная почка). Эти морфологический изменения характеризуют так называемый амилоиднолипоидный нефроз, а правильнее — нефротическую стадию амилоидоза почек.
Диабетический гломерулосклероз (смотри полный свод знаний Гломерулосклероз диабетический) представляет собой одно из наиболее ярких проявлений диабетической микроангиопатии. В основе его лежит пролиферация мезангиальных клеток в ответ на засорение гломерулярного фильтра и мезангия, а также повышенное образование клетками мембраноподобного вещества. Склероз капиллярных петель может носить диффузный или очаговый характер, что послужило основанием для выделения диффузной, узловатой и смешанной форм диабетического гломерулосклероза. Гломерулосклероз нередко дополняется экссудативными проявлениями диабетической нефропатии в виде «фибриновых колпачков» на капиллярных петлях и «капсульной капли», а также гликогенной «инфильтрацией» эпителия узкого сегмента нефрона, где происходит полимеризация глюкозы в гликоген.
Парапротеинемический нефроз (синонимы: миеломная нефропатия, миеломная почка), развивающийся в связи с наличием парапротеинемии и парапротеинурии, характеризуется прежде всего нарастающей дистрофией (гиалиново-капельная, вакуольная) и гибелью эпителия канальцев преимущественно проксимального сегмента, обилием цилиндров и кристаллов белка в канальцах, что ведёт к их обструкции, нарастающему нефрогидрозу, лимфостазу и повышению внутрипочечного давления. Как реакция на эти изменения возникает склероз и гиалиноз стромы, восходящий от пирамид к корковому веществу почек, что завершается перигломерулярным склерозом и нарастающей гибелью нефронов. Иногда к этим изменениям присоединяется параамилоидоз.
Симптоматика и течение
Жалобы больных — слабость, анорексия, жажда, сухость во рту, отеки, ощущение тяжести в поясничной области.
Отеки развиваются быстро, сопровождаясь олигурией, и могут достигать степени анасарки, сочетаться с водянкой полостей (асцит, гидроторакс, гидроперикард), но могут и отсутствовать. При больших отёках на бледной коже появляются полосы растяжения, признаки дистрофии кожи и её дериватов — волос, ногтей: шелушение, сухость, ломкость. При нарастании гидроторакса и гидроперикарда появляется одышка при физической нагрузке и в покое. При отсутствии асцита удаётся пальпировать увеличенную печень мягкоэластической консистенции. Тоны сердца могут быть приглушены, при анемии возникает тахикардия и систолический шум. По мере уменьшения отёков выявляется атрофия скелетной мускулатуры. Функция щитовидной железы может быть снижена. К этим клинические, признакам добавляются проявления основного заболевания, что крайне отягощает состояние больного.
По характеру течения выделяют три варианта невротического синдрома: эпизодический, появляющийся лишь в начале основного заболевания с исходом в ремиссию или рецидивирующий, чередующийся с ремиссиями (функция почек при этом в течение 10—20 лет сохраняется нормальной); персистирующий, когда Нефротический синдром сохраняется, несмотря на лечение, в течение 4—8 лет без снижения функций почек (соответствует прежнему понятию «хронический нефроз»); прогрессирующий с переходом за 1 — 3 года в стадию хронической почечной недостаточности. Вариант течения в определённой мере зависит от нозологической формы Нефротический синдром и морфологический особенностей нефропатии. Так, эпизодическое течение свойственно аллергическому Нефротический синдром; быстропрогрессирующее течение, помимо экстракапиллярного первичного гломерулонефрита, наблюдается при фокально-сегментарном гломерулярном гиалинозе. У лиц пожилого возраста чаще встречаются второй и третий варианты течения.
Осложнения
При Нефротический синдром развиваются разнообразные и многочисленные осложнения: отёк мозга, сетчатки глазного дна, нефротический криз (гиповолемический шок), флеботромбозы, вторичная инфекция, инфаркт миокарда, инсульт головного мозга, острая почечная недостаточность и другие В период лекарственной терапии количество осложнений может резко увеличиться, так как некоторые препараты при Нефротический синдром могут оказывать токсическое, аллергическое действие, а также провоцировать отдельные симптомы Нефротический синдром (например, в период стероидной терапии может усилиться гиперкоагуляция и развиться тромбоз).
Диагноз
Диагноз при ярко выраженной клинические, симптоматике Нефротический синдром не вызывает затруднений. Важное значение в диагностике имеют лабораторный методы исследования. Наиболее частый лабораторный признак при Нефротический синдром— большая протеинурия (смотри полный свод знаний). Количество белка иногда достигает 20—50 грамм/суток. Белки, определяемые в моче, плазменного происхождения, однако с противоположным соотношением по молекулярному весу: в моче — максимальное количество альбумина, относительно увеличено содержание α1 и β-глобулинов и понижено (иногда до следов) α2-и γ-глобулинов. Состав белков мочи и селективность протеинурии зависят от характера основного заболевания. Неселективный характер протеинурии, то есть выделение высокомолекулярных белков, отражает большую тяжесть поражения нефрона. Однако не селективность протеинурии может быть обратимой.
Выделение с мочой больших количеств таких ферментов, как трансамидиназа, лейцинаминопептидаза, кислая фосфатаза, (β-глюкуронидаза, N-ацетилглюкозоаминидаза и другие свидетельствует об остром процессе в почках, о тяжести поражения нефрона, особенно эпителия извитых канальцев, о высокой проницаемости клеточных мембран. Кроме того, в моче больных с Нефротический синдром определяют до 5 электрофоретических фракций гликопротеидов и 2—3 фракции липопротеидов. Характерна для Нефротический синдром и гипераминоацидурия, особенности которой зависят скорее от основного заболевания (смотри полный свод знаний Аминоацидурия).
Гипопротеинемия (смотри полный свод знаний Протеинемия) — постоянный симптом Нефротический синдром Общий белок крови снижается до 4,0 и даже 3,0 грамм/100 миллилитров, в связи с чем онкотическое давление плазмы падает с 30—40 до 10—15 сантиметров водного столба. В происхождении этого признака, помимо потери белков с мочой, имеет значение ещё и усиленный катаболизм их (в частности, альбумина), перемещение некоторых белков во внеклеточную жидкость, потеря их через отёчную слизистую оболочку кишечника, снижение синтеза белков в печени и так далее Диспротеинемия, неизменно сопутствующая гипопротеинемии, выражается в резком уменьшении концентрации альбумина в сыворотке крови, повышении α2 и β-глобулиновых фракций. Содержание гамма-глобулинов чаще снижено, хотя при некоторых заболеваниях может быть и повышено. В α2-глобулиновой фракции повышено содержание гаптоглобинов и α2-макроглобулина. При этом повышается и содержание фибриногена, синтез которого находится в прямой зависимости от количества гаптоглобина.
При значительно выраженном Нефротический синдром изменяется соотношение в сыворотке крови содержания основных классов иммуноглобулинов: снижаются иммуноглобулины классов А и G и повышается уровень иммуноглобулинов класса М. Уровень сывороточного комплемента (по гемолитической активности) снижается в разной мере — более резко при волчаночном Нефротический синдром, реже — при Нефротический синдром амилоидного происхождения.
Гиперлипидемия — также типичный признак Нефротический синдром Проявляется повышенным содержанием холестерина, триглицеридов и фосфолипидов, дислипопротеидемией (смотри полный свод знаний Липопротеиды). Увеличивается концентрация пребета и бета-липопротеидов при нормальном или пониженном количестве альфа-липопротеидов. Гиперлипидемия связана с рядом причин: задержкой липопротеидов как высокомолекулярных веществ в сосудистом русле, усиленным синтезом холестерина в печени, снижением активности липолитических ферментов (липопротеидлипазы), возможно, нарушением метаболической функции почек. В тесной связи с гиперлипидемией находится липидурия, которая определяется по наличию жировых цилиндров в моче, иногда жира, лежащего свободно или внутри слущённого эпителия.
Кроме того, при Нефротический синдром наблюдается гиперкоагуляция крови — от небольшой степени активации свёртывающей системы крови до предтромботического состояния и криза локальной или диссеминированной внутрисосудистой коагуляции. Этим нарушениям гемостаза (смотри полный свод знаний) способствует состояние депрессии системы фибринолиза и снижение антикоагулянтной активности крови. Лишь в крайне редких случаях при Нефротический синдром удаётся наблюдать высокую фибринолитическую активность. Факторами, способствующими гиперкоагуляции, являются снижение уровня таких ингибиторов протеиназ, как антитромбин-III, альфаантитрипсин; при повышении уровня главного антиплазмина — альфа2-макроглобулина, а также усиление адгезивных свойств тромбоцитов. Отмечаются электролитные сдвиги в сыворотке крови (снижение концентрации кальция, калия), гиповитаминоз (особенно недостаточность витаминов С и D), изменения содержания микроэлементов. Гуморальные нарушения сказываются на метаболизме и функциональный состоянии лейкоцитов крови. Так, в лимфоцитах крови снижается активность окислительно восстановительных ферментов (сукцинат- и альфа-глицерофосфат — дегидрогеназ), в нейтрофилах изменяется активность щелочной и кислой фосфатаз.
У многих больных выявляется анемия, гипертромбоцитоз и ускоренная РОЭ.
В мочевом осадке, помимо эритроцитов, могут определяться в значительном количестве лимфоциты (10—60%). Наряду с гиалиновыми цилиндрами при Нефротический синдром обнаруживают и восковидные, что соответствует большой протеинурии.
Дифференциальный диагноз основан главным образом на данных биопсии почки и других органов и тканей (кожи, десны, слизистой оболочки прямой кишки, печени), а также пункции грудины (при подозрении на миеломную болезнь). Имеют значение и некоторые лабораторный методы (анализ на LE-клетки и титр антител к ДНК при подозрении на системную красную волчанку и так далее).
Лечение
Необходимы ранняя госпитализация, быстрая дифференциальная диагностика с попыткой воздействия на инициальные и ведущие механизмы основного заболевания.
Назначается бессолевая, богатая калием диета с содержанием животного белка 1 грамм/килограмм веса больного. Большие белковые нагрузки приводят к росту протеинурии и угнетению фибринолитической системы крови.
Учитывая гипоальбуминемию, при Нефротический синдром суточные дозы лекарственных препаратов должны быть полуторными или двойными, распределёнными для приёма дробно; при выраженном отёке их лучше вводить внутривенно.
Стероидная терапия показана при Нефротический синдром лекарственной, волчаночной этиологии, мембранозном гломерулонефрите.
Цитостатики (имуран, циклофосфамид или лейкеран) назначают больным с Нефротический синдром, имеющим противопоказания для стероидной терапии или при её неэффективности. Отмечен особенно хороший эффект в результате их применения при лечении Нефротический синдром у больных узелковым периартериитом, синдромом Вегенера. Их нередко назначают в сочетании с кортикостероидами. Антикоагулянты (гепарин 20—50 тысяч ЕД в 1 суток в течение 4—6 недель, нередко в сочетании с курантилом, иногда с непрямыми антикоагулянтами) показаны и эффективны при всех нозологических и морфологический формах Нефротический синдром, при которых выражен механизм внутрисосудистой коагуляции.
Противовоспалительные средства (индометацин, бруфен) показаны для лечения больных мембранозным и мезангиопролиферативным гломерулонефритом с Нефротический синдром
Из симптоматических средств при Нефротический синдром употребляют мочегонные (салуретики, антагонисты альдостерона), дозы которых подбирают индивидуально. Хороший эффект можно ожидать при сочетании лазикса с раствором обессоленного альбумина или реополиглюкина внутривенно. При лечении резистентных отёков у больных с олигурией могут быть применены ультрафильтрация (смотри полный свод знаний) и гемофильтрация (смотри полный свод знаний).
Рекомендуется санаторно-курортное лечение (Байрам-Али, Бухара, а в период ремиссии — курорты Южного берега Крыма). Показания к курортному лечению зависят от основного заболевания и степени его активности.
Прогноз
Прогноз при Нефротический синдром зависит от многих факторов: возраста, характера основного заболевания, особенностей нефропатии, морфологический особенностей (ремиссий не бывает при фибропластическом варианте), длительности Нефротический синдром, клинические, формы, адекватности лекарственной терапии и другие
Причины смертельных исходов: прогрессирующее течение основного заболевания и (или) нефропатии как основного проявления этого заболевания; почечная недостаточность (острая или хроническая); инфекционные осложнения (пневмония, эмпиема плевры, сепсис, апостематозный нефрит); тромбоэмболии; агранулоцитоз; желудочно-кишечные кровотечение и другие
Профилактика
Меры специфической профилактики развития Нефротический синдром не разработаны. Определённое значение могут иметь раннее и успешное лечение заболеваний, осложняющихся Нефротический синдром, а также диспансеризация больных.
Врождённый (семейный) нефротический синдром
Врождённый (семейный) нефротический синдром объединяет группу заболеваний, при которых отеки появляются в первые недели жизни ребёнка в связи с развитием у него изменений в почках ещё в антенатальном периоде. Нефротический синдром иногда имеет семейный характер и нередко наследуется. Наибольшее распространение Нефротический синдром отмечалось в Финляндии (заболеваемость среди новорожденных до 1980 год составляла 1 на 10 000 родившихся). В других странах, в том числе и в СССР, заболевание встречается значительно реже.
Особенности клинико-морфологических проявлений Нефротический синдром у детей в Финляндии дало основание для выделения так называемый врождённого Нефротический синдром финского типа, представляющего собой генетически детерминированный вариант патологии, наследуемый по аутосомно-рецессивному типу. Предполагается, что мутация впервые произошла около 400 лет тому назад в одном из северно-западный районов Финляндии, который в течение многих лет имел характерные черты изолята, где были нередки родственные браки. Рождению ребёнка с Нефротический синдром предшествует тяжело протекающая беременность. При этом выявляются иммунологический феномены несовместимости между матерью и плодом (в крови у матери и ребёнка обнаружены преципитирующие антитела, направленные против антигенов почек плода и плаценты). Роды нередко преждевременные, плацента увеличена и составляет более 25% веса тела новорожденного.
Врождённый Нефротический синдром финского типа проявляется с первых дней жизни ребёнка (реже — после 2 месяцев) и характеризуется выраженными отёками, протеинурией, тяжёлой гипопротеинемией с резкой гипогаммаглобулинемией. Такие дети отстают в физическом развитии, у них выражены стигмы дизэмбриогенеза (деформация ушных раковин, синдактилия, гипертелоризм, грыжи и другие); они гипотрофичны, а динамичны, подвержены инфекционным болезням и другим заболеваниям, сопровождающимся септическими осложнениями, которые, как правило, являются причиной летального исхода.
При гистологический исследовании почек выявляются чёткообразные цепочечные расширения проксимальных отделов нефрона (псевдокистоз), обнаруживаются различной степени выраженности гломерулярные, тубулярные и интерстициальные изменения, степень которых нарастает по мере прогрессирования заболевания, а также большое количество фетальных гломерул и гломерул с увеличенным диаметром.
Врождённый Нефротический синдром, встречающийся у детей спорадически (в других странах), выявляется, как правило, в более позднем возрасте (нередко в конце первого или на втором году жизни), течение его более лёгкое. В отличие от врождённого Нефротический синдром финского типа в почках при этом могут наблюдаться следующие варианты морфологический изменений: мезангиальный диффузный склероз, фокальный или сегментарный гиалиноз и гломерулонефрит с экстрамембранной локализацией па то л. процесса; микрокистоз наблюдается реже.
Диагноз врождённого Нефротический синдром не представляет трудности и основан на данных анамнеза, типичной клинические, картине, данных лабораторных исследований и биопсии почки.
Лечение не разработано. Применение глюкокортикоидных гормонов и иммунодепрессантов неэффективно и нередко утяжеляет течение синдрома. Уменьшение анасарки иногда достигается применением диуретиков. В Финляндии было проведено несколько операций по пересадке почки детям до одного года с Нефротический синдром, но они не дали положительных результатов.
Прогноз неблагоприятный. Дети погибают от интеркуррентных заболеваний или почечной недостаточности.
Профилактика не разработана. Имеются данные о возможности антенатальной диагностики путём определения а-фетопротеина в амниотической жидкости. В случае положительной реакции рекомендуется прерывание беременности.
Экспериментальный нефротический синдром
Модели Нефротический синдром позволяют уточнить его патогенетические механизмы и воспроизвести ряд изменений почек, характерных для этого синдрома.
Адекватной моделью первичного Нефротический синдром считают аминонуклеозидный нефроз. Эта модель морфологически наиболее близка липоидному нефрозу, так как, по данным Фаркар (М. G. Farquhar) и Дж. Пелейда, основные изменения при введении аминонуклеозида возникают в эпителии гломерулярного фильтра: подоциты теряют малые отростки, вакуолизируются, в цитоплазме их появляется большое количество белковых гранул; повреждается щелевидная мембрана. Кефалидес, Форселл-Нотт (N. A. Kefalides, L. Forsell-Knott) установили, что базальная мембрана гломерулярного фильтра изменяется вторично, теряет гидроксилизин, гидроксипролин и глицин; электронно-микроскопически находят изменения её макромолекулярной структуры. Она становится повышенно проницаемой для крупномолекулярных белковых частиц (каталаза, ферритин).
Моделями вторичного Нефротический синдром можно считать экспериментальные грамм ломерулонефрит и амилоидоз, а также поражения почек, возникающие у подопытных животных под воздействием некоторых органических и неорганических веществ. Для воспроизведения в эксперименте гломерулонефрита, сопровождающегося Нефротический синдром, используют разные воздействия: однократное или повторное парентеральное введение гетеро или гомологичного белка, сенсибилизацию чужеродным белком и создание условий локализации гиперергической реакции (инфекции) в почках, микроорганизмы и их токсины, а также смеси бактериальных антигенов с гомологичной почечной тканью, антипочечную цитотоксическую сыворотку, гомологичную или аутологичную ткань почки. Эти эксперименты позволили доказать роль иммунологический повреждения (циркулирующие иммунные комплексы, антипочечные антитела) базальной мембраны клубочковых капилляров в развитии Нефротический синдром Циркулирующие иммунные комплексы находят в таких случаях электронно-микроскопически на эпителиальной стороне базальной мембраны; при повреждении её антителами обнаруживают характерные изменения, подобные тем, которые возникают при пневморенальном синдроме Гудпасчера.
При экспериментальном амилоидозе, для воспроизведения которого обычно вводят казеин, показана роль глубоких обменных нарушений (белка, липидов) в развитии Нефротический синдром Классические проявления амилоидного Нефротический синдром (или нефроза) в виде мочевого синдрома, гипопротеинемии, гиперлипидемии, отёков обнаруживают на 6—8-й недель опыта (нефротическая стадия), когда амилоид «загружает» не только пирамиды, но и клубочки, а дистрофия канальцев и лимфостаз достигают максимума. В основе развития Нефротический синдром в этих случаях, по мнению В. В. Серова, лежит первичное повреждение амилоидом гломерулярного фильтра и вторичная недостаточность тубулолимфатического аппарата реабсорбции почек. На моделях повреждения почек некоторыми органическими и особенно неорганическими (ртуть, свинец, уран) соединениями Д. С. Саркисовым, П. И. Ремезовым показано значение токсических воздействий и состояния почечных канальцев для развития Нефротический синдром Эти модели более применимы для изучения механизмов острой почечной недостаточности.
|
Игнатова М.С.; Полянцева Л.P.; Серов В.В. |

|
⇐ Перейти на главную страницу сайта |
⇑ Вернуться в начало страницы ⇑ |
Библиотека Ordo Deus ⇒ |
⇐ Нефросклероз |
⇓ Полный свод знаний. Том первый А. ⇓ |
Ниманна — Пика болезнь ⇒ |
|
Все статьи в полном изложении, Вы можете найти в большой медицинской энциклопедии — Главный редактор: академик АН СССР (РАН) и АМН СССР (РАМН) Б.В. Петровский. — Москва издательство «Советская энциклопедия» 1989г. |
|
Внимание! Вы находитесь в библиотеке «Ordo Deus». Все книги в электронном варианте, содержащиеся в библиотеке «Ordo Deus», принадлежат их законным владельцам (авторам, переводчикам, издательствам). Все книги и статьи взяты из открытых источников и размещаются здесь только для чтения. |
|
Вся информация на сайте Ordo Deus находится в свободном доступе. Ordo Deus не предоставляет информацию на платной основе. |
|
Все авторские права сохраняются за правообладателями. Если Вы являетесь автором данного документа и хотите дополнить его или изменить, уточнить реквизиты автора, опубликовать другие документы или возможно вы не желаете, чтобы какой-то из ваших материалов находился в библиотеке, пожалуйста, свяжитесь с нами по e-mail: |
Вас категорически не устраивает перспектива безвозвратно исчезнуть из этого мира? Вы не желаете закончить свой жизненный путь в виде омерзительной гниющей органической массы пожираемой копошащимися в ней могильными червями? Вы желаете вернувшись в молодость прожить ещё одну жизнь? Начать всё заново? Исправить совершённые ошибки? Осуществить несбывшиеся мечты? Перейдите по ссылке: «главная страница».
|
© Ordo Deus, 2010. При копировании ссылка на сайт http://www.ordodeus.ru обязательна. |