Геморрагические диатезы |
||
 |
 |
|
Оглавление
|
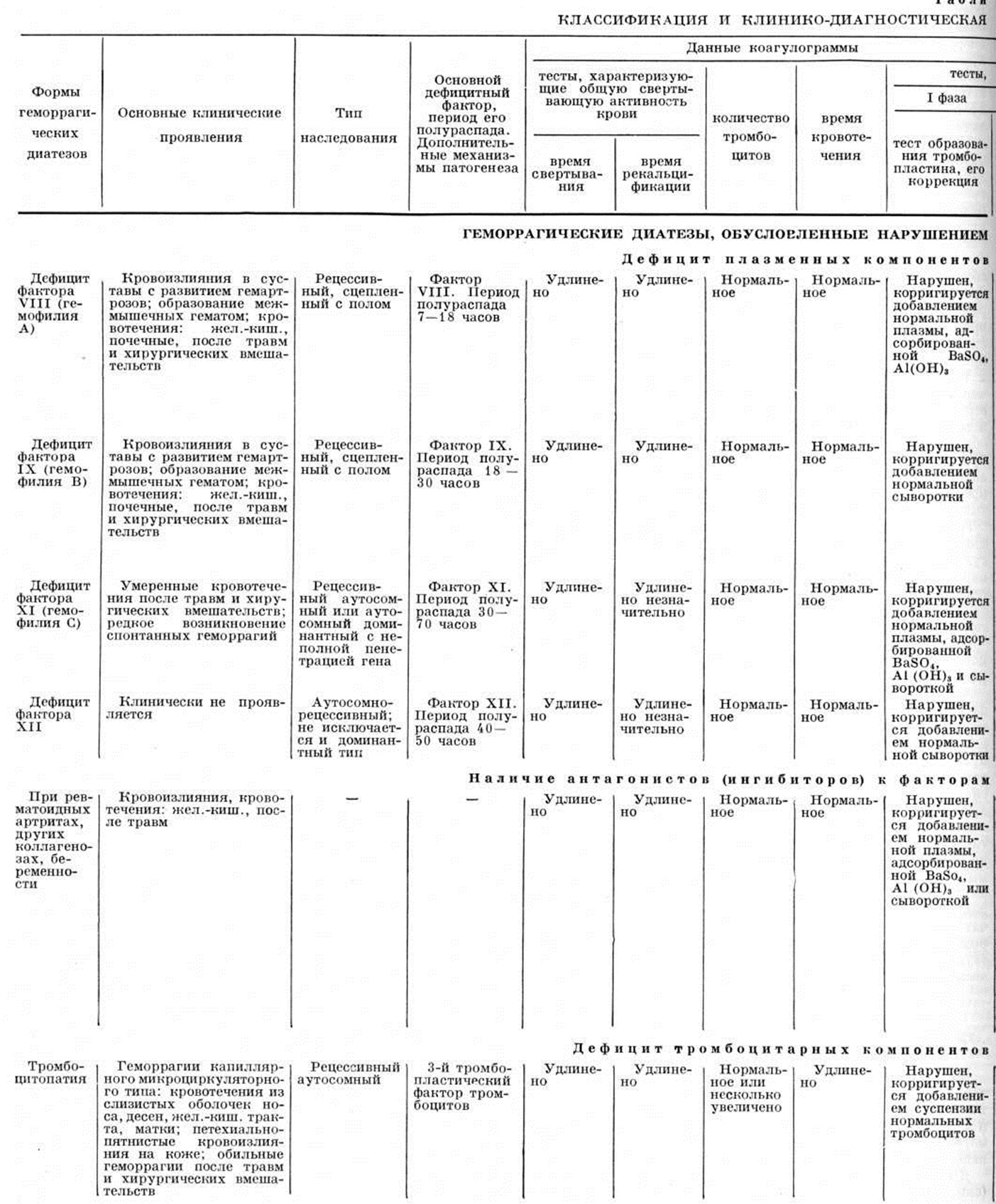
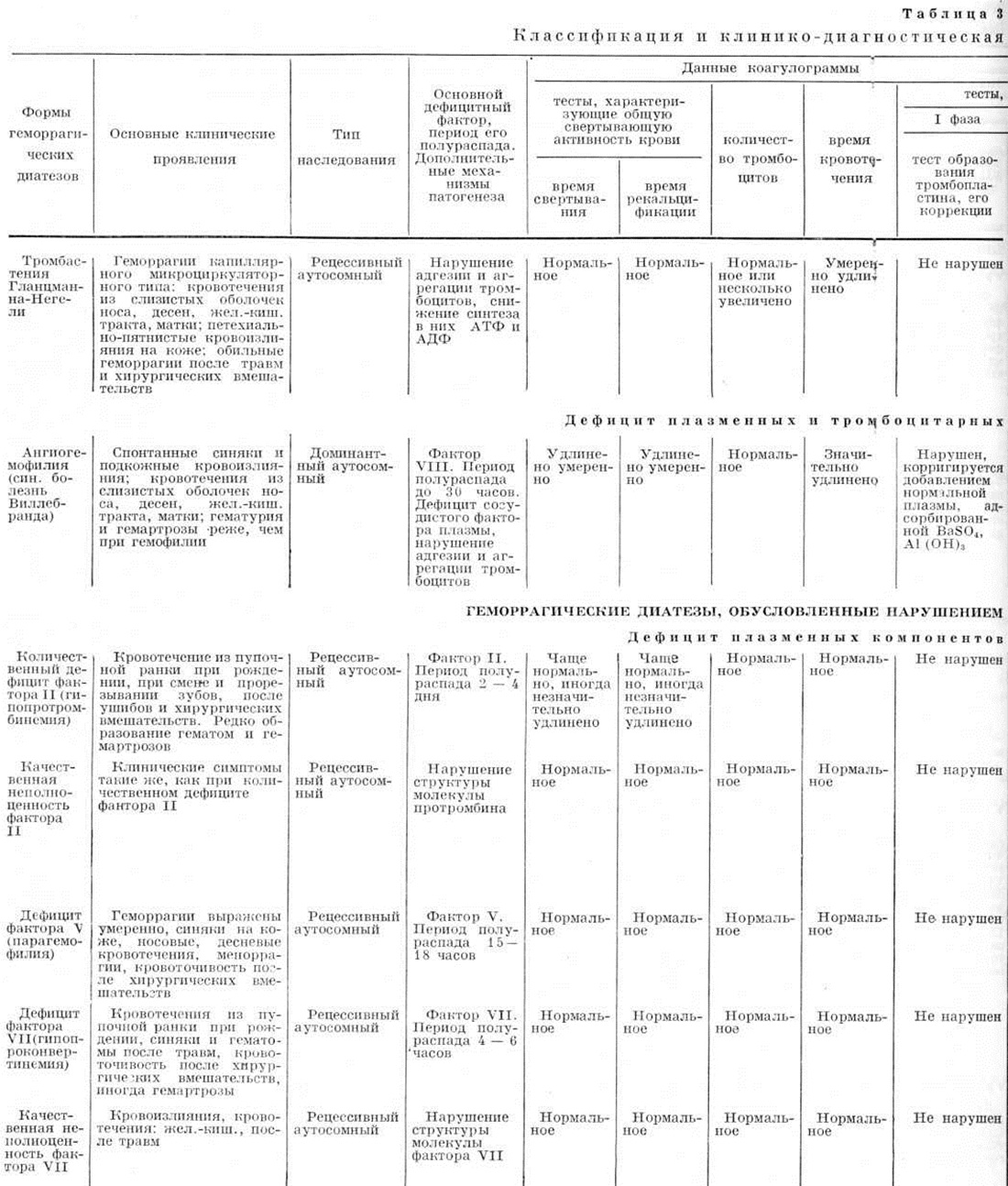
Геморрагические диатезыГеморрагические диатезы (греческий haimorrhagia кровотечение; диатезы) — группа наследственных и приобретённых болезней, основным клиническим признаком которых является повышенная кровоточивость — наклонность организма к повторным кровотечениям и кровоизлияниям, самопроизвольным или после незначительных травм. Механизм развития Геморрагические диатезы разнообразен и может быть связан с патологией различных компонентов свёртывающей системы крови (смотри) — плазменных и тромбоцитарных, усилением фибринолиза (смотри), наличием диссеминированного внутрисосудистого свёртывания, циркулирующих в крови антикоагулянтов; повышением проницаемости сосудов или аномалией сосудистой стенки. Каждый из указанных механизмов может быть первичным (Геморрагические диатезы как самостоятельное заболевание) либо сопровождать другие заболевания (симптоматический Геморрагические диатезы). Первичные Геморрагические диатезы относят к врождённым семейно-наследственным заболеваниям, характерный признак которых — дефицит какого-либо одного фактора свёртывания крови; исключением является болезнь Виллебранда, при которой нарушаются несколько факторов гемостаза — фактор VIII, сосудистый фактор, адгезивность тромбоцитов. Симптоматические Геморрагические диатезы характеризуются недостаточностью нескольких факторов свёртывания крови. Классификация. В основу рабочей классификации Геморрагические диатезы может быть положена схема нормального процесса свёртывания крови. Заболевания сгруппированы соответственно фазам процесса свёртывания крови. Геморрагические диатезы, обусловленные нарушением первой фазы свёртывания крови (образование тромбопластина): Дефицит плазменных компонентов тромбопластинообразования — фактора VIII (гемофилия А), фактора IX (гемофилия В), фактора XI (гемофилия С), фактора XII. Наличие антагонистов (ингибиторов) факторов VIII и IX. Дефицит тромбоцитарных компонентов тромбопластинообразования — количественная недостаточность тромбоцитов (первичная и симптоматическая), качественная недостаточность тромбоцитов (тромбоцитопатии). Ангиогемофилия (синонимы болезнь Виллебранда). Геморрагические диатезы, обусловленные нарушением второй фазы свёртывания крови (образование тромбина): Дефицит плазменных компонентов тромбинообразования — фактора II (протромбина), фактора V (Асглобулина), фактора VII (проконвертина), фактора X (фактора Стюарта — Прауэр). Наличие антагонистов (ингибиторов) тромбинообразования. Наличие ингибиторов к факторам II, V, VII и X. Геморрагические диатезы, обусловленные нарушением третьей фазы свёртывания крови (образование фибрина): дефицит плазменных компонентов фибринообразования — фактора I (фибриногена), количественный и качественный дефицит фактора XIII (фибринстабилизирующего фактора). Геморрагические диатезы, обусловленные ускоренным фибринолизом. Геморрагические диатезы, обусловленные развитием диссеминированного внутрисосудистого свёртывания: синдром дефибринации (синонимы: тромбогеморрагический синдром, диссеминированное внутрисосудистое свёртывание, коагулопатия потребления). Геморрагические диатезы, обусловленные нарушением первой фазы свёртывания кровиДефицит плазменных компонентов тромбопластинообразования — факторов VIII, IX, XI и XII. Дефицит факторов VIII и IX — смотри Гемофилия. |
Дефицит фактора XI (синонимы: гемофилия С, дефицит предшественника тромбопластина плазмы, синдром Розенталя) впервые описали в 1953 г. Розенталь (R. L. Rosenthal), Дрескин и Розенталь (О. Н. Dreskin, N. Rosenthal). В последующие 10 лет было описано св. 120 больных во всех частях света, однако статистических данных о распространённости дефицита фактора XI нет. Наследуется по аутосомно-доминантному типу с неполной пенетрацией гена; не исключается и аутосомнорецессивный характер наследования. Обнаруживается у лиц обоего пола с одинаковой частотой. Фактор XI — предшественник тромбопластина плазмы, активизируется под действием активного фактора XII, способствует превращению фактора IX в активную форму; при недостаточности его нарушается образование тромбопластина. Это белок, при электрофорезе мигрирует в зоне β2глобулинов. Стабилен при хранении, не потребляется в процессе свёртывания крови. Место синтеза не установлено.
Симптомы заболевания напоминают гемофилию. Кровоточивость выражена умеренно: обычно кровотечения после травм и небольших хирургических вмешательств (экстракция зубов, тонзиллэктомия и другие). Спонтанные геморрагии возникают редко. Трудоспособность больных не нарушается.
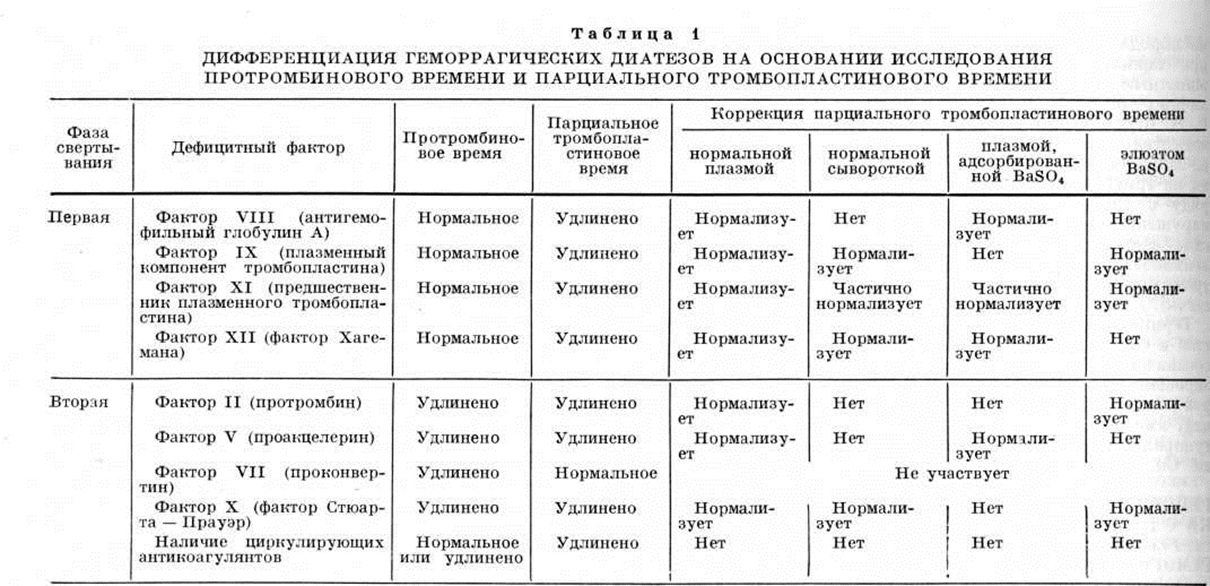
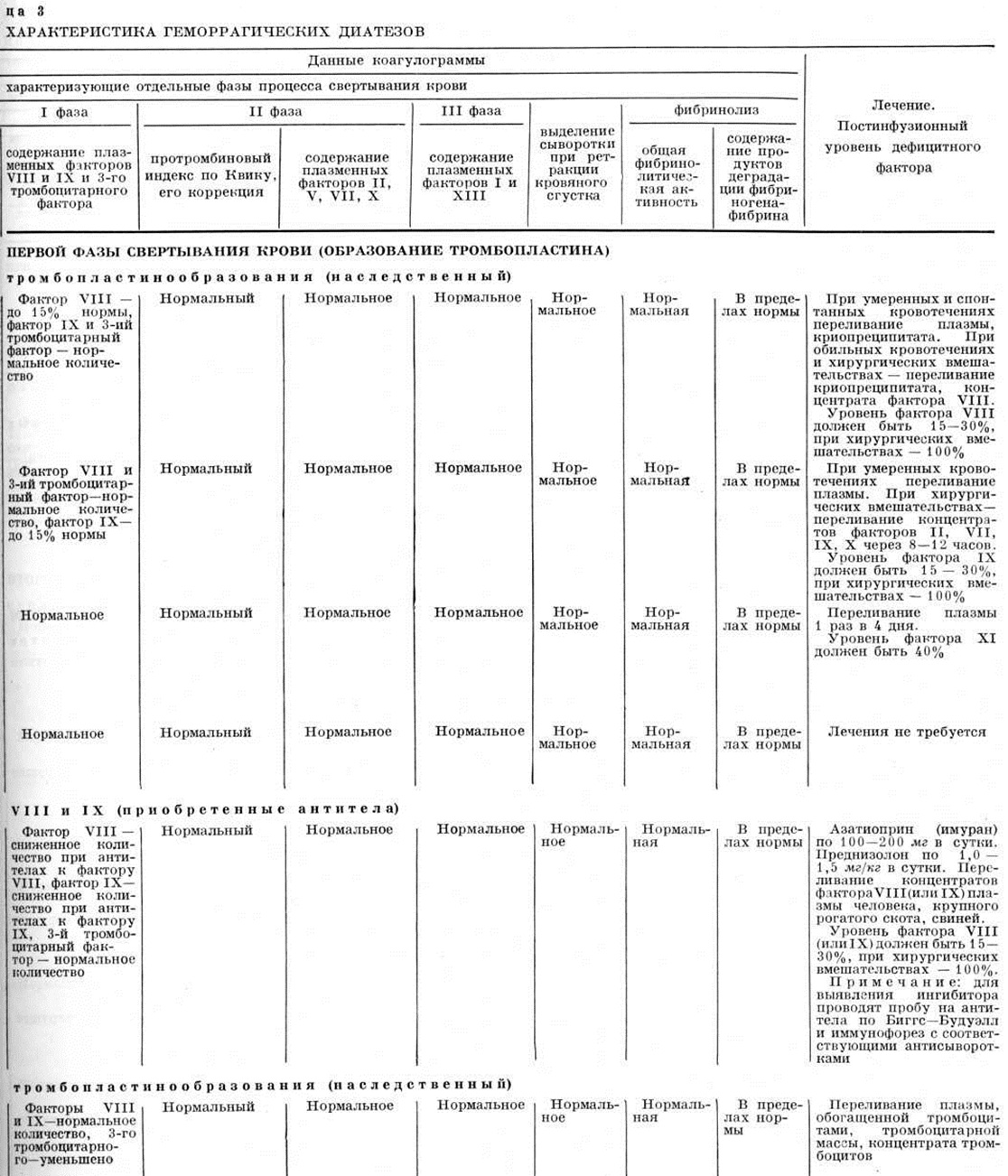
Диагноз ставят на основании снижения уровня фактора XI ниже 20%, а также характерных данных коагулограммы (смотри): некоторое увеличение времени свертывания крови и времени рекальцификации, нарушение теста потребления протромбина, образования тромбопластина (по Биггс — Дугласу) и парциального тромбопластинового времени (таблица 1) при нормальном содержании факторов VIII и IX плазмы и 3-го фактора тромбоцитов.
Кровотечение останавливают путём тампонады, прижатия кровоточащего участка. В редких случаях обильных кровотечений хороший эффект даёт переливание плазмы.
Дефицит фактора XII впервые описали в 1955 год Ратнов и Копли (О. D. Ratnoff, A. L. Copley). К 1970 год зарегистрировано более 100 больных. Дефицит фактора XII наследуется по аутосомно-рецессивному типу; доминантный характер наследования полностью не исключается.
Фактор XII (синонимы: фактор контакта, фактор Хагемана) является глюкопротеином. В плазме находится в неактивной форме, активируется при контакте с чужеродной поверхностью. При электрофорезе мигрирует с 3-глобулинами, стабилен при нагревании до t× 56×. Активирует фактор XI и способствует агрегации тромбоцитов.
Дефицит фактора XII клинически не проявляется. Диагноз ставится только по данным коагулограммы: удлинение времени свёртывания в силиконированных пробирках и на силиконированных стёклах, нарушение парциального тромбопластинового времени (нормализуется добавлением нормальной или адсорбированной BaSO4 плазмы и сыворотки) при нормальном протромбиновом времени (таблица 1).
Лечения больных обычно не требуется; прогноз благоприятный.
 |
 |
 |
Наличие в крови антагонистов (ингибиторов) к факторам VIII и IX.
Ингибиторами фактора VIII являются антитела к фактору VIII, которые относят к иммуноглобулинам класса IgG, IgM. В 1940 г. Лознер (Е. L. Lozner) и соавторами описали у больных с заболеванием, напоминающим гемофилию, наличие антикоагулянта. Последний был обнаружен также у больных гемофилией, которые получали множественные трансфузии, что и явилось доказательством принадлежности этих ингибиторов к антителам.
Приобретённые ингибиторы к фактору VIII описаны при ревматизме, острой красной волчанке, лейкозах, сепсисе и других заболеваниях, а также в поздние сроки беременности и после родов.
Симптомы заболевания клинически напоминают гемофилию, развиваются в любом возрасте на фоне основного заболевания; семейный анамнез не отягощён. Диагноз ставят на основании данных коагулограммы (удлинение времени свёртывания крови, уменьшение потребления протромбина, нарушение теста образования тромбопластина, уменьшение фактора VIII, положительная проба Биггс — Бидуэлл на антитела к фактору VIII) и подтверждают методом иммуноэлектрофореза (появляется дуга преципитации против специфической антисыворотки).
Лечение должно быть направлено на основное заболевание, на подавление продукции антител и купирование геморрагий. С целью подавления продукции антител назначают иммунодепрессанты — азотиоприн (имуран) по 100—200 миллиграмм и преднизолон по 1—1,5 миллиграмм/килограмм ежедневно до полного исчезновения антител. Из гемостатических сред более эффективно переливание концентратов фактора VIII, особенно гетерогенных, но последние антигенны и их можно применять только при обильных, длительных кровотечениях, угрожающих жизни; повторное введение гетерогенных препаратов может дать тяжёлые посттрансфузионные реакции. Прогноз зависит от основного заболевания и тяжести геморрагического синдрома. Он значительно ухудшается при кровоизлияниях в жизненно важные органы (головной мозг, мышца сердца и другие).
Ингибиторы фактора I X описаны как у больных гемофилией В, так и при других состояниях. Принципы диагностики, методы лечения и прогноз такие же, как при ингибиторах фактора VIII.
Дефицит тромбоцитарного компонента тромбопластинообразования развивается вследствие количественной недостаточности тромбоцитов при тромбоцитопенической пурпуре (смотри Пурпура тромбоцитопеническая), симптоматической тромбоцитопении (смотри Гипопластическая анемия, Лейкозы) и качественной неполноценности тромбоцитов (тромбопатии).
С момента описания Гланцманном (Е. Glanzmann, 1918) тромбастении обнаружен ряд заболеваний, причиной которых является качественная неполноценность тромбоцитов. Классификация этих заболеваний представляет большие трудности. Браунштейнер (Н. Braunsteiner, 1955) предлагает делить их на тромбопатии и тромбастении. Термином «тромбопатия» он обозначает недостаточность в тромбоцитах фактора 3 (тромбопластического), под термином «тромбастения» — недостаточность в тромбоцитах фактора 8 (фактор ретракции). С накоплением новых сведений стало ясно, что качественная недостаточность тромбоцитов является комплексной. Следовательно, классификация по одному признаку может привести к ошибкам. По решению Международного комитета по гемостазу и тромбозам более удачным признан термин «тромбопатия» или «тромбоцитопатия». В эту группу относят любую качественную недостаточность тромбоцитов: уменьшение содержания в них отдельных факторов или недостаточное освобождение этих факторов в процессе свёртывания крови (смотри Тромбоцитопатии).
Ангиогемофилия — семейно-наследственная форма Геморрагические диатезы, обусловленная врождённым дефицитом в плазме антигеморрагического сосудистого фактора Виллебранда и фактора VIII. Основным лабораторным тестом является удлинение времени кровотечения (до 1 часа и более); количество тромбоцитов, индекс ретракции кровяного сгустка и время свёртывания крови нормальные (смотри Ангиогемофилия).
Геморрагические диатезы, обусловленные нарушением второй фазы свёртывания крови
Дефицит плазменных компонентов тромбинообразования — факторов II, V, VII и X.
Врождённая количественная недостаточность фактора II (протромбина) — истинная гипопротромбинемия; описана Роудсом и Фитц-Хью (J. Е. Rhoads, Jr. Т. Fitz-Hugh, 1941) под названием идиопатической гипопротромбинемии у больного с тяжёлыми кровотечениями (протромбиновое время резко удлинено, другие факторы протромбинового комплекса — V, VII, X — не были исследованы). В 1947 год Квик (A. J. Quick) описал у двух братьев выраженную кровоточивость, удлинение протромбинового времени и нормальный уровень фактора V, а в 1955 год — значительное уменьшение протромбина у девочки. Заболевание встречается редко. Описано около 20 больных с достоверной гипопротромбинемией [Силер (R. A. Seeler), 1972]. Наследуется по аутосомно-рецессивному типу. Болеют лица обоего пола.
|
 |
|
Рис | ||
Протромбин превращается в тромбин под действием активного фактора X. Протромбин (фактор II) — глюкопротеин мигрирует при электрофорезе с α2-глобулином. Стабилен при хранении и нагревании, растворим в воде. Период полураспада протромбина 12—24 часа. Синтезируется в печени при участии витамина К. 75—85% протромбина потребляется во время свёртывания (смотри Протромбин).
Клинически наблюдаются признаки повышенной кровоточивости, которые появляются иногда в момент рождения в виде кровотечения из пупочного канатика, позже при прорезывании и смене зубов, у больных женщин — с началом менструаций. Возникают носовые кровотечения, меноррагии, кровотечения после родов, ушибов, удаления зубов, хирургических вмешательств (тонзиллэктомий и другие). Могут появляться межмышечные гематомы и гемартрозы, обычно без нарушения функций суставов. Гематурия, желудочно-кишечного кровотечения наблюдаются редко. С возрастом кровоточивость уменьшается, несмотря на то, что дефицит протромбина остаётся.
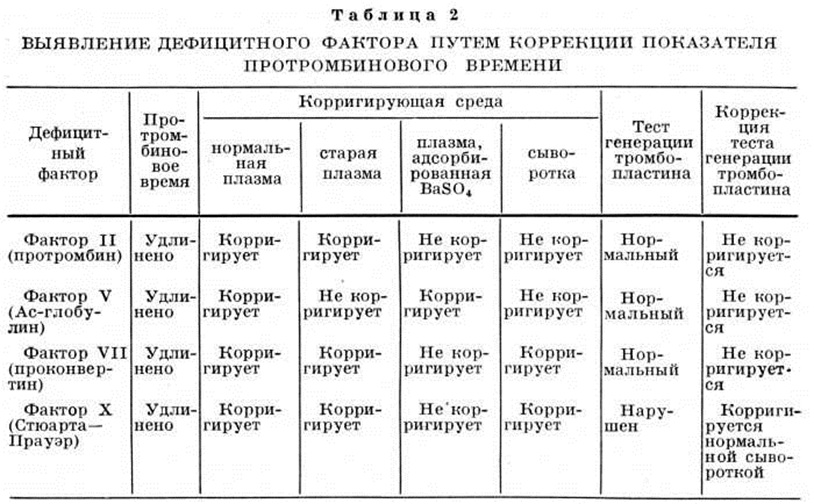
Диагноз устанавливают на основании данных коагулограммы: снижение протромбинового индекса по Квику и при определении двухступенчатым методом (смотри Протромбиновое время), коррекция протромбинового времени по Квику нормальной свежей и «старой» плазмой, сохранение дефицита протромбина после добавления сыворотки и адсорбированной плазмы (таблица 2).
Нарушение парциального тромбопластинового времени нормализуется добавлением нормальной плазмы и элюата BaSO4 (таблица 1).
Лечение при кровотечениях проводят переливанием плазмы или крови. При больших хирургических вмешательствах предпочтительнее переливать концентраты дефицитного фактора, вводя PPSB — препарат, содержащий протромбин, проконвертин, фактор Стюарта—Прауэр, фактор IX (смотри Гемофилия, антигемофильные препараты). Для гемостаза достаточно, чтобы уровень протромбина в результате трансфузий составлял 40% нормы.
Прогноз зависит от степени дефицита фактора II; при появлении кровоизлияний в жизненно важные органы прогноз значительно ухудшается.
Качественная недостаточность протромбина (диаспротромбия) описана Шапиро (S. S. Shapiro) с соавторами (1969) и Джоссо (Е. Josso) с соавторами (1972), которые обнаружили у членов одной семьи заболевание с клинической, признаками гипопротромбинемии. Тип наследования аутосомно-рецессивный. Уровень протромбина составлял 15—10% нормы (определение одно и двухступенчатым методами).
При исследовании со стафиллокоагулазой и методом иммуноэлектрофореза со специфическими антисыворотками к протромбину человека содержание протромбина было нормальным.
Симптомы заболевания, методы лечения и прогноз такие же, как и при врождённой количественной недостаточности протромбина.
Симптоматическая недостаточность протромбина наблюдается при заболеваниях с нарушением функции печени, при лечении антикоагулянтами непрямого действия (производными кумарина), при дефиците витамина К, при синдроме диссеминированного внутрисосудистого свёртывания. В коагулограмме, кроме снижения уровня протромбина, выявляют недостаточность тех факторов свёртывания крови, которые синтезируются главным образом в печени (факторы I, V, VII). Лечение должно быть направлено на купирование кровоточивости. Назначают переливания плазмы, при развитии анемии переливают кровь. С целью повышения синтеза протромбина применяют витамин К в инъекциях и викасол. При передозировке антикоагулянтов непрямого действия к этим препаратам добавляют рутин в дозе до 0,1 грамм 3 раза в сутки и немедленно отменяют антикоагулянт. Обязательным является лечение основного заболевания, успешность которого определяет прогноз.
Недостаточность фактора V (синонимы гипопроакцелеринемия).
Фактор V (синонимы Ас-глобулин) ускоряет превращение протромбина в тромбин активированным фактором X. Это белок, при электрофорезе мигрирует между β и γ-глобулинами; лабилен: быстро разрушается при хранении и нагревании. Период полураспада короткий (12—15 часов). Полностью потребляется при свёртывании крови и в сыворотке не обнаруживается. Синтезируется в печени при участии витамина К.
Парагемофилия — наследственная недостаточность фактора V, впервые описана в 1947 год Овреном (P. A. Owren) и Квиком. Заболевание встречается редко, точной статистики нет. По данным Силера, к 1972 год описано 58 больных (30 мужчин и 28 женщин). Наследуется заболевание по аутосомно-рецессивному типу; некоторые авторы допускают доминантный тип наследования. Болезнь обычно встречается в семьях, где имеются браки между родственниками.
Симптомы заболевания могут появиться в момент рождения. Течение болезни обычно более лёгкое, чем при дефиците других факторов протромбинового комплекса. У большинства больных обнаруживают кровоизлияния в кожу, носовые кровотечения. Глубокие межмышечные гематомы и гемартрозы образуются редко. У женщин часто бывают меноррагии. Описывают кровотечения после оперативных вмешательств, экстракции зубов, после родов. Диагноз устанавливают на основании данных коагулограммы: снижение протромбинового индекса, который корригируется добавлением адсорбированной BaSO4 плазмы, лишённой факторов II и VII. Нарушение парциального тромбопластинового времени нормализуется добавлением нормальной плазмы и плазмы, адсорбированной BaSO4 (таблица 2). Иногда дефицит фактора V сочетается с уменьшением активности фактора VIII. Эти случаи необходимо дифференцировать с гемофилией А (смотри Гемофилия), ангиогемофилией (смотри).
Лечение: заместительная трансфузия свежей плазмы или крови; при обильных кровотечениях и больших хирургических вмешательствах трансфузию повторяют каждые 6—8 часов, для гемостаза достаточно поддерживать содержание фактора V в пределах 10—30% нормы. Концентратов фактора V не получено.
Прогноз зависит от частоты и длительности кровотечений и локализации геморрагий: значительно ухудшается при кровоизлияниях в головной мозг. Полное выздоровление невозможно. Иногда в зрелом возрасте кровоточивость уменьшается при сохранении дефицита фактора V.
Симптоматическая недостаточность фактора V возникает на фоне заболеваний, осложнённых поражением печени (гепатиты, циррозы печени, лейкозы и другие). Клинической, признаки болезни определяются основным заболеванием, к ним присоединяются геморрагические проявления различной тяжести и локализации. Приобретённый дефицит фактора V всегда сочетается с недостаточностью других факторов свёртывания (I, II, VII, X), что с учётом анамнеза позволяет дифференцировать это состояние от врождённого дефицита фактора V.
Лечение должно включать активную терапию основного заболевания; с гемостатической целью проводят трансфузии плазмы или крови.
Недостаточность фактора VII может быть наследственной и симптоматической (смотри Гипопроконвертинемия).
Наследственный дефицит фактора X (фактора Стюарта — Прауэр) описали Квик и Хасси (С. V. Hussey, 1953): у больной отмечалось умеренное удлинение протромбинового времени и нарушение потребления протромбина.
В 1956 год Тельфер (Т. P. Telfer) с соавторами опубликовал результаты исследования аналогичной больной с двойным дефектом, который они обозначали как дефицит фактора Прауэр, а Хофи (С. Houghie) с соавторами независимо от них описал сходное заболевание у мужчины, которое обозначили как недостаточность фактора Стюарта. Впоследствии была показана идентичность этих факторов, и указанный дефицит был назван болезнью Стюарта—Прауэр. Заболевание встречается относительно редко. К 1972 год было описано около 25 наблюдений. Тип наследования — аутосомно-рецессивный.
Фактор X активирует переход протромбина в тромбин. Является белком, мигрирует при электрофорезе в зоне α1-глобулинов. Синтезируется в печени. Период полураспада 30—70 часов. Стабилен при хранении и быстро разрушается при нагревании; не потребляется в процессе свёртывания крови; обнаруживается и в плазме, и в сыворотке. При его дефиците нарушаются I и II фазы процесса свёртывания крови.
Клинически недостаточность фактора X редко проявляется геморрагиями. Только при почти полном его отсутствии возникают носовые кровотечения, меноррагии, кровотечения из слизистых оболочек желудочно-кишечного тракта и почек, внутричерепные кровоизлияния, гемартрозы и межмышечные гематомы. Содержание фактора X может увеличиваться при беременности и поэтому во время родов кровотечения, как правило, отсутствуют. Однако в послеродовом периоде наблюдаются тяжёлые кровотечения, что связано с падением концентрации фактора X. После хирургических вмешательств, выполненных без соответствующей подготовки, также возможны кровотечения.
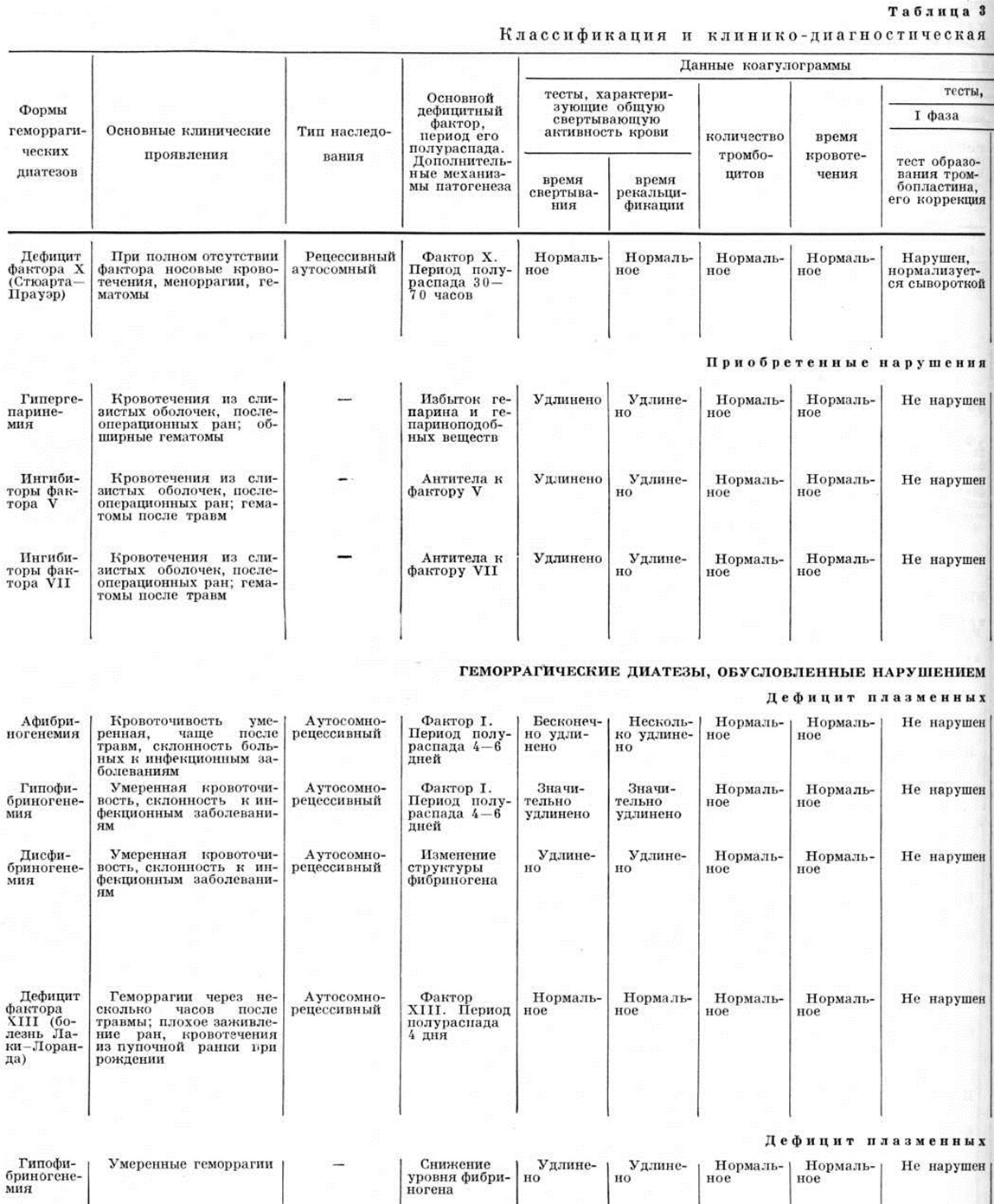
Диагноз основывается на данных коагулограммы: потребление протромбина уменьшено, тест образования тромбопластина нарушен и нормализуется добавлением нормальной плазмы и сыворотки, парциальное тромбопластиновое время удлинено и нормализуется добавлением нормальной плазмы, сыворотки, а также элюата BaSO4 (таблица 3).
Протромбиновое время удлинено, корригируется добавлением нормальной и «старой» плазмы и сыворотки (таблица 2). Дифференцируют с Геморрагические диатезы, обусловленными недостаточностью других факторов протромбинового комплекса (II, V и VII) и с гемофилией. При дефиците факторов II и V протромбиновое время нормализуется добавлением нормальной свежей плазмы, добавление сыворотки не изменяет этого времени, тест образования тромбопластина не нарушен. При дефиците фактора VII протромбиновое время корригируется добавлением нормальной плазмы (свежей и консервированной) и нормальной сыворотки. Использование змеиного яда Расселла в тесте одноступенчатого протромбинового времени вместо тромбопластина способствует дифференциации дефицита факторов VII и X: при недостаточности фактора VII протромбиновое время нормализуется, при дефиците фактора X — остаётся удлинённым. Тест образования тромбопластина не нарушен при недостаточности фактора VII; при дефиците фактора X тест образования тромбопластина нарушен за счёт сывороточного компонента (нормализуется при добавлении нормальной сыворотки). Дефицит фактора X дифференцируют с гемофилией на основании нормального протромбинового времени при нарушенном тесте образования тромбопластина.
Лечение направлено на остановку спонтанных кровотечений. С целью повышения уровня фактора X (необходимо повысить более чем на 10%) переливают плазму; при операциях и в послеродовом периоде более эффективна трансфузия концентратов PPSB и его аналогов.
Прогноз зависит от степени дефицита фактора X, частоты и локализации геморрагий.
Наличие антагонистов (ингибиторов) тромбинообразования.
Антагонисты тромбина. Под термином «антитромбин» подразумевают общую способность плазмы или сыворотки нейтрализовать тромбин. Различают антитромбин I, II, III, IV, V и VI.
Антитромбином I является фибрин (смотри), который адсорбирует тромбин после свёртывания крови, что имеет большое значение для прекращения дальнейшего свёртывания крови при наступившем гемостазе. При лизисе фибрина тромбин освобождается.
Антитромбин II — гепарин (смотри) содержится в высоких концентрациях в печени, лёгких, мышцах. Растворим в воде, осаждается спиртом, ацетоном и кислотой. Относится к мукополисахаридам, молекулярный вес 10 000—12 000. Гепарин и его Ко-фактор предотвращает превращение протромбина в тромбин.
Антитромбин III вызывает необратимое разрушение тромбина в плазме. При фракционировании выделяется с альбумином, при электрофорезе мигрирует с α2глобулином, разрушается под действием эфира, нагреванием до t° 80° и при pH выше 9,5 и ниже 6,0. Молекулярный вес 64 000. Избыток антитромбина III ведёт к повышенной кровоточивости.
Антитромбин IV появляется во время свёртывания крови. Его значение в развитии повышенной кровоточивости не установлено.
Антитромбин V обнаружен в крови больных, страдающих ревматоидными артритами. Может обусловливать повышенную кровоточивость у этой группы больных.
Антитромбин VI описан впервые Ковальским (Е. Kowalski, 1959). Он образуется при частичном лизисе фибриногена и уменьшает влияние тромбина и полимеризацию фибрин-мономера; разрушается при нагревании в течение 20 минут до t° 60°, не диализируется; при электрофорезе мигрирует между β и γ-глобулинами, не адсорбируется BaSO4, осаждается 50% сульфатом аммония.
Среди антагонистов тромбина наибольшее значение имеет гепарин (смотри).
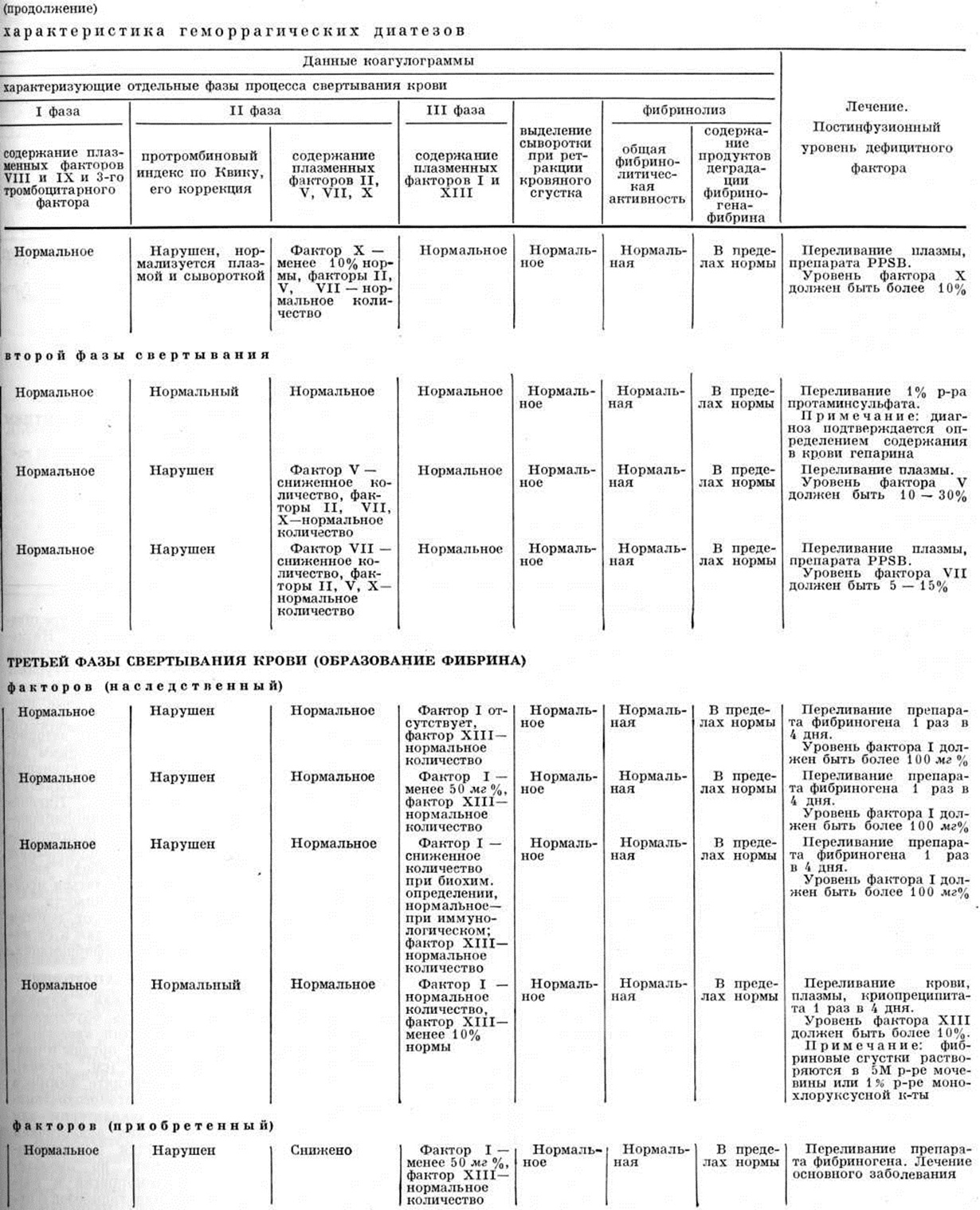
Гипергепаринемия бывает чаще приобретённой, но может быть и врождённой. Она развивается при коллагенозах, лейкозах, передозировке гепарина (при лечении тромбоэмболических осложнений), при операциях с экстракорпоральным кровообращением, анафилактическом шоке и другие Симптомы гипергепаринемии характеризуются бурными кровотечениями из слизистых оболочек, послеоперационных разрезов и ран, обширными и глубокими гематомами. Диагноз основывается на данных коагулограммы: удлинение времени свёртывания крови и тромбинового времени, которые корригируются добавлением протамин-сульфата или толуидинового синего (проба Сирмаи). Дифференцируют с Геморрагические диатезы, обусловленными наличием приобретённых антител к различным факторам свёртывания. При последних время свёртывания крови также удлинено, но оно не нормализуется при добавлении протамин-сульфата и толуидинового синего. При наличии антител к фактору VIII нарушается тест потребления протромбина и тест образования тромбопластина, обнаруживают положительную пробу Биггс—Бидуэлл; при наличии антител к фактору VII удлинено протромбиновое время и время свёртывания крови.
Лечение сводится к внутривенному введению 1% раствора протамин-сульфата, количество вводимого препарата зависит от степени гипергепаринемии; контроль за лечением заключается в определении уровня гепарина в крови.
Прогноз зависит от течения основного заболевания и тяжести геморрагического синдрома.
Антагонисты факторов протромбинового комплекса (II, V, VII, X) возникают у больных с врождённой недостаточностью этих факторов или при заболеваниях, протекающих с нарушениями в иммунокомпетентной системе (коллагенозы, бронхиальная астма, диспротеинемии). Клинической, признаки аналогичны наблюдаемым при гипопротромбинемии. Диагноз основывается на данных коагулограммы: уменьшение содержания одного из факторов протромбинового комплекса с помощью однои двухступенчатого методов определения протромбина и подтверждается результатами иммунофореза со специфическими антисыворотками.
Геморрагические диатезы, связанные с нарушением третьей фазы свертывания крови (образование фибрина)
Дефицит плазменных компонентов фибринообразования. Недостаточность фибриноген а (фибриногенемия и гипофибриногенемия) — смотри А фибриногенемия, недостаточность фактора XIII.
Дефицит фактора XIII (синонимы болезнь Лаки—Лоранда) впервые описан Дуккертом (F. Duckert, 1960). Статистика не разработана. Наследуется по аутосомно-рецессивному типу, не исключается и наследование, сцепленное с полом.
Фактор XIII (синонимы: фибриназа, фибринстабилизирующий фактор, фибринолигаза) участвует в стабилизации фибрина: превращает растворимый фибрин S (soluble) в стабильный фибрин I (insoluble). Содержится в крови в неактивной форме, активируется тромбином в присутствии ионов кальция. Стабилен при хранении, частично термостабилен. Период полураспада 4 дня.
Геморрагии возникают при уменьшении в крови фактора XIII (ниже 10%). Характерно позднее возникновение кровоточивости — через несколько часов после травмы; описаны обширные гематомы, синяки, желудочно-кишечного кровотечения, кровотечения из пупочной ранки. Вследствие дефицита фактора XIII плохо заживают раны (рыхлость сгустка препятствует прорастанию его фибробластами).
Диагноз основывается на типичной клинике (позднее возникновение кровотечений и плохое заживление ран) и данных коагулограммы: тесты, характеризующие систему гемостаза, не нарушаются. При исследовании растворимости сгустка (в пятимолярном растворе мочевины или 1% растворе монохлоруксусной кислоты) обнаруживается его нестабильность.
Лечение необходимо при выраженной кровоточивости или при проведении этим больным хирургических вмешательств. Применяют трансфузии цельной крови, плазмы, а в тяжёлых случаях криопреципитата. Для эффективного гемостаза достаточно повышение уровня фактора XIII (более 10%). Прогноз обычно благоприятный.
|
 |
|
|
 |
|
|
 |
|
|
 |
|
|
 |
|
|
 |
|
|
 |
|
|
 |
|
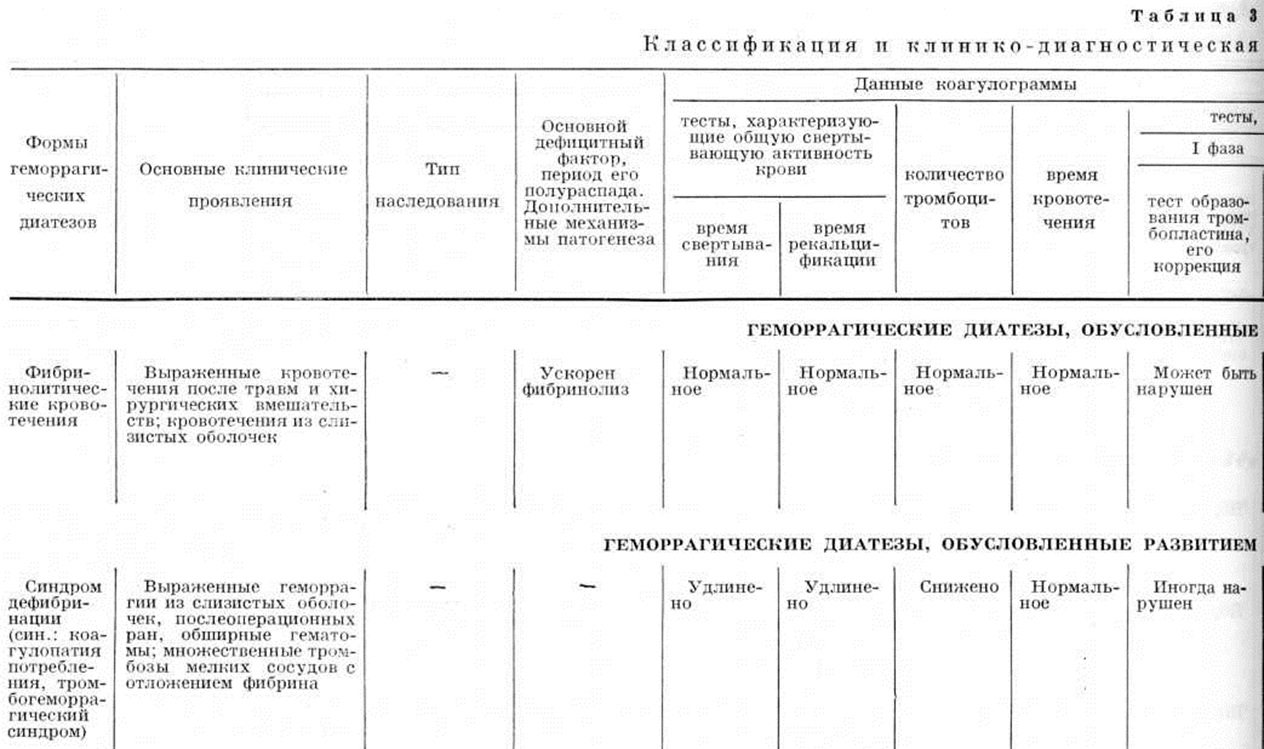
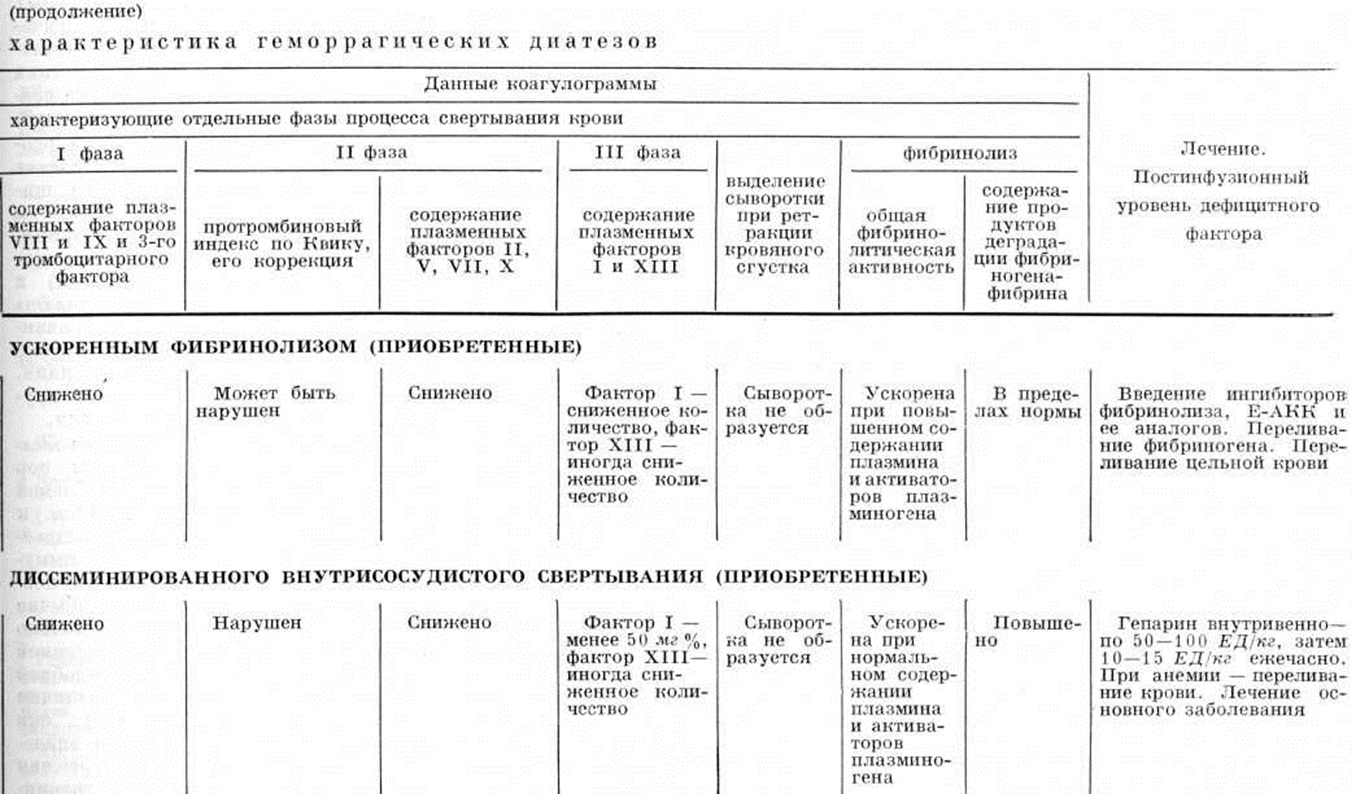
Геморрагические диатезы, обусловленные ускоренным фибринолизом
Процессы фибринолиза ускоряются вследствие повышения синтеза плазмина или недостаточного синтеза антиплазмина (смотри Фибринолиз).
Геморрагические диатезы, обусловленные развитием диссеминированного внутрисосудистого свёртывания.
Синдром дефибринации (синонимы: коагулопатия потребления, тромбогеморрагический синдром) развивается на фоне клиники метастазирующей злокачественной опухоли, внутрисосудистого гемолиза, шока, ожоговой болезни, при преждевременной отслойке плаценты, внутриутробной смерти плода, при попадании в кровяное русло веществ с тромбопластической активностью.
Бленвилль (Н. М. D. Blainville, 1834) обнаружил, что внутривенное введение животным мозговой ткани ведёт к немедленной гибели их в результате массивного внутрисосудистого свёртывания крови. Вулдридж (L. С. Wooldridge, 1886) нашёл, что медленное внутривенное введение животным тканевого тромбопластина не ведёт к гибели животного, проявляясь развитием состояния несвертываемости крови. Обата (J. Obata, 1919) наблюдал, как инъекции тромбопластических веществ вызывают образование тромбов в мелких кровеносных сосудах. Миллс (С. A. Mills, 1921) выявил при этом уменьшение концентрации фибриногена. По данным Мелланби (J. Mellanby, 1933) и Уорнера (Е. D. Warner) с соавторами (1939), аналогичный эффект наблюдался при внутривенном введении тромбина. Вайнер (А.Е. Weiner) с соавторами (1950), Шнайдер и Пейдж (С. L. Schneider, Е. W. Page, 1951) высказали предположение, что при попадании в кровяное русло тромбопластических веществ происходит внутрисосудистое свёртывание, в результате которого истощаются запасы фибриногена и потребляются факторы свёртывания. Джексон (D. P. Jackson) с соавторами (1955) обнаружили у таких больных гипофибриногенемию, уменьшение количества тромбоцитов и концентрации протромбина. Аналогичный механизм был установлен и для синдрома дефибринации при внутривенном введении тромбопластических веществ [Копли, 1945; Ратнов и Конли (С. L. Conley); Шнайдер, 1957]. Симптомы заболевания проявляются развитием интенсивного внутрисосудистого свёртывания крови (фаза гиперкоагулемии). В процессе массивного внутрисосудистого свёртывания крови используются все прокоагулянты (коагулопатия потребления): снижается уровень факторов I, II, V, VII, VIII, XIII и количество тромбоцитов (фаза гипокоагулемии). Вследствие гиперкоагуляции и отложения фибрина в сосудах активируется фибринолитическая система (фаза вторичного фибринолиза и дефибринации), что сопровождается увеличением продуктов деградации фибриногена и фибрина при нормальном уровне активаторов плазминогена и плазмина. Синдром дефибринации по течению может быть острым, подострым и хроническим. Острое течение синдрома дефибринации длится несколько часов или дней и часто остаётся нераспознанным. Наблюдается при шоке, внутрисосудистом гемолизе, ожоговой болезни, хирургических вмешательствах (на лёгких, поджелудочной железе и другие), в акушерской практике (при отслойке плаценты, внутриутробной смерти плода), септическом аборте, острых вирусных инфекциях и других состояниях.
Геморрагии проявляются в виде петехий на коже, кровотечений и гематом после инъекций и разрезов. Особенно обильные кровотечения развиваются при дефибринации на фоне акушерской патологии.
Подострое течение синдрома дефибринации длится в течение нескольких недель. Чаще возникает при метастазирующих злокачественных опухолях, лейкозах, внутриутробной смерти плода. Кровоточивость может быть генерализованной и локальной, что обусловлено локальной травмой или распадом очага поражения (напр., опухоль желудка). В некоторых случаях ведущими симптомами являются тромбозы вен и артерий.
Хронический течение синдрома дефибринации обычно наблюдается при сосудистой патологии (гигантские гемангиомы — синдром Казабаха — Мерритта, массивные каверноматозные изменения в сосудах, особенно в системе селезёночной и воротной вен). Кровоточивость и тромбозы выражены слабо или отсутствуют.
Диагноз ставят на основании клиники и данных коагулограммы: тромбоцитопения, удлинение тромбинового времени, снижение уровня фибриногена, недостаточность факторов II, V, VIII, повышение содержания продуктов деградации фибриногена и фибрина при нормальном содержании плазмина и активаторов фибринолиза. Дифференцируют с приобретённой гипофибриногенемией у больных с тяжёлыми заболеваниями печени, к-рая может сопровождаться уменьшением факторов II, V, VII и X, но содержание фактора VIII остаётся нормальным. При первичном фибринолизе наряду со снижением содержания фибриногена и факторов II, V, VII, VIII и X повышается уровень плазмина и его активаторов. При наличии циркулирующих антикоагулянтов уровень фибриногена и других факторов свёртывания обычно не снижается, нет активации фибринолиза.
При синдроме дефибринации прежде всего необходимо лечение основного заболевания, на фоне которого он развился. Для купирования геморрагий некоторые авторы полагают обоснованным введение антикоагулянтов прямого действия. Обычно вводят внутривенно гепарин: начальная доза — 50—100 ЕД на 1 килограмм веса; затем ежечасно по 10—15 ЕД на 1 килограмм. Внутримышечное введение его не рекомендуется, так как из-за замедленного его всасывания трудно проконтролировать наступление гипергепаринемии. Однако это мнение разделяется не всеми исследователями. При сочетании синдрома дефибринации с резкой тромбоцитопенией дозу гепарина уменьшают вдвое, одновременно назначая переливание крови и фибриногена. Назначение гепарина при отсутствии синдрома дефибринации усугубляет кровоточивость и может нанести вред больному. Кумариновые препараты применяют для длительного лечения, но, чтобы затормозить дефибринацию, необходимы высокие дозы, которые, резко уменьшая содержание факторов свёртывания, усиливают кровотечения. Ингибиторы фибринолиза (Σ-аминокапроновая кислота и её аналоги) противопоказаны, так как они ведут к образованию внутрисосудистых тромбов, введение их может сопровождаться прогрессированием кровоточивости.
Прогноз зависит как от течения основного заболевания, так и от интенсивности синдрома дефибринации.
Патологическая анатомия
Патологоанатомическая картина при Геморрагические диатезы может быть обусловлена остаточными явлениями кровоизлияний (смотри) в различные органы и признаками малокровия (смотри Анемия). При вторичном дефиците факторов свёртывания крови патологоанатомические изменения характерны для основного заболевания; аналогичная картина наблюдается и при синдроме дефибринации, но преобладают признаки геморрагий в различных органах или тромбозов с отложением фибрина в сосудах, особенно мелких.
Осложнения
Осложнения при Геморрагические диатезы зависят от локализации геморрагий. При повторных кровоизлияниях в суставы возникают гемартрозы (смотри); при образовании обширных гематом в области прохождения крупных нервных стволов возможно сдавление нервов с развитием параличей, парезов (смотри); при кровоизлияниях в головной мозг появляются симптомы, характерные для нарушения мозгового кровообращения (смотри). При повторных переливаниях крови и плазмы может развиться сывороточный гепатит. У больных с полным отсутствием факторов свёртывания возможно образование антител, что значительно уменьшает эффективность трансфузий; возможны посттрансфузионные реакции. Обнаружено образование антител к эритроцитарным, лейкоцитарным и тромбоцитарным антигенам, что осложняет проведение переливаний и требует специального подбора доноров.
Профилактика
Профилактика рецидивов состоит в переливании соответствующих трансфузионных сред, которые повышают уровень дефицитного фактора и купируют геморрагии. Большое значение имеют медико-генетические консультации, ориентирующие супругов из семей с врождённой патологией в системе свёртывания крови в отношении планирования потомства.
Геморрагические диатезы у детей
Среди госпитализируемых в стационары детей с болезнями системы крови около половины составляют больные с Геморрагические диатезы
Распространённость Геморрагические диатезы имеет определённую возрастную зависимость. Наследственные формы Геморрагические диатезы проявляются, как правило, с рождения или вскоре после рождения, например, гипо- и афибриногенемии (смотри), врождённые тромбоцитопатии (смотри), синдром Вискотта — Олдрича (смотри Вискотта — Олдрича синдром) и другие. Приобретённые формы Геморрагические диатезы чаще наблюдаются в дошкольном и школьном возрасте, например пурпура тромбоцитопеническая (смотри), геморрагический васкулит (смотри Шенлейна — Геноха болезнь) и другие.
Транзиторная недостаточность факторов свёртывания крови получила название геморрагической болезни новорожденных. Она проявляется в первые дни жизни кровоизлияниями в кожу, мышцы, слизистые оболочки (петехии, экхимозы, гематомы), в мозг, кровотечениями из слизистых оболочек желудочно-кишечного тракта (мелена, кровавая рвота), пупочной ранки и так далее.
Основной причиной геморрагической болезни новорожденных (особенно недоношенных) является низкое содержание некоторых факторов свёртывания крови (проконвертина, протромбина и другие) и повышенное содержание веществ, обладающих антикоагулянтной активностью (антитромбопластин, антитромбины, в первую очередь гепарин, фибринолизин и другие), на фоне свойственной этому периоду детства повышенной проницаемости сосудистой стенки. Транзиторная недостаточность связана также с незрелостью отдельных органов (особенно печени), с недостаточностью витамина К. У некоторых новорожденных с гемолитической болезнью повышенная кровоточивость объясняется наличием антиэритроцитарных антител, трансплацентарно перешедших от матери, обладающих групповой антигенной активностью к тромбоцитам ребёнка: у больного обнаруживается поэтому не только анемия, но и тромбоцитопения (смотри Гемолитическая болезнь новорожденных). Интеркуррентные и инфекционные заболевания, асфиксия и нарушения метаболизма (особенно ацидоз) у новорожденных с дефицитом факторов свёртывания крови значительно усиливают кровоточивость. Веккио и Бушар (F. Vecchio, Bouchard) описали особый вид Геморрагические диатезы у новорожденных, возникающий после 8-го дня жизни, иногда через несколько недель, и характеризующийся внезапностью появления и тяжестью кровоточивости, сопровождающейся дефицитом компонентов протромбинового комплекса, а также других плазменных факторов свёртывания крови (IX, X и др.) при отсутствии функционального поражения печени. Патогенетическая связь этой формы Геморрагические диатезы с авитаминозом подтверждается эффективностью парентерального введения витамина К. Возникновение этих поздних идиопатических форм Геморрагические диатезы связано, по-видимому, с потерей способности гепатоцитов использовать витамин К, который всасывается из желудочно-кишечного тракта нормально. Этот вид Геморрагические диатезы следует отличать от гиповитаминоза К, обусловленного холестазом или поражением тонкой кишки.
Лечение основано на патогенетических механизмах нарушения гемостаза. При наследственных формах используются средства, устраняющие дефицит отдельных факторов свёртывания крови, а также средства, подавляющие антикоагулянтную активность крови.
В профилактике наследственных форм Геморрагические диатезы большое значение имеют медико-генетические консультации, а приобретённых — предупреждение заболеваний, способствующих их возникновению.
|
Мазурин А.В.; Орлова Л.Д. |

|
⇐ Перейти на главную страницу сайта |
⇑ Вернуться в начало страницы ⇑ |
Библиотека Ordo Deus ⇒ |
⇐ Гемоперитонеум |
⇓ Полный свод знаний. Том первый А. ⇓ |
Геморрагические лихорадки ⇒ |
|
Все статьи в полном изложении, Вы можете найти в большой медицинской энциклопедии — Главный редактор: академик АН СССР (РАН) и АМН СССР (РАМН) Б.В. Петровский. — Москва издательство «Советская энциклопедия» 1989г. |
|
Внимание! Вы находитесь в библиотеке «Ordo Deus». Все книги в электронном варианте, содержащиеся в библиотеке «Ordo Deus», принадлежат их законным владельцам (авторам, переводчикам, издательствам). Все книги и статьи взяты из открытых источников и размещаются здесь только для чтения. |
|
Библиотека «Ordo Deus» не преследует никакой коммерческой выгоды. |
|
Все авторские права сохраняются за правообладателями. Если Вы являетесь автором данного документа и хотите дополнить его или изменить, уточнить реквизиты автора, опубликовать другие документы или возможно вы не желаете, чтобы какой-то из ваших материалов находился в библиотеке, пожалуйста, свяжитесь с нами по e-mail: |
Вас категорически не устраивает перспектива безвозвратно исчезнуть из этого мира? Вы не желаете закончить свой жизненный путь в виде омерзительной гниющей органической массы пожираемой копошащимися в ней могильными червями? Вы желаете вернувшись в молодость прожить ещё одну жизнь? Начать всё заново? Исправить совершённые ошибки? Осуществить несбывшиеся мечты? Перейдите по ссылке: «главная страница».
|
© Ordo Deus, 2010. При копировании ссылка на сайт http://www.ordodeus.ru обязательна. |