Амиотрофии (БМЭ) |
||
 |
 |
|
Оглавление
|
Амиотрофии (БМЭ)Амиотрофии (amyotrophia;rpeеское отрицательная приставка a-, mys, myos — мышца и trophe — питание) — нарушение трофики мышц вследствие поражения периферического мотонейрона, сопровождающееся их дегенеративно-дистрофическими изменениями, истончением и нарушением сократительной функции. Различают группу наследственно обусловленных амиотрофий, среди которых выделяют спинальные амиотрофии и невральные. Амиотрофия может быть также результатом поражения мышц при коллагенозах, интоксикациях, инфекционно-паразитарных заболеваниях, эндокринных нарушениях, рефлекторных расстройствах и других (смотри Атрофия мышечная). Амиотрофии обусловлены вовлечением в патологический процесс клеток передних рогов спинного мозга,а также их отростков и спинномозговых нервов и характеризуются постепенным развитием параличей иннервируемых мышц, качественной реакцией перерождения мышц, снижением их электровозбудимости. Атрофии подвергаются как саркоплазма, так и миофибриллы. Атрофия мышечного волокна, возникающая в результате нарушения его иннервации, называется денервационной, вторичной амиотрофией, в отличие от первичного атрофического процесса в мышцах, при котором функция двигательного нейрона не страдает (смотри Миопатия). Гистологически при неврогенных повреждениях выявляется пучковый характер распределения атрофии мышечных волокон, сохранение наряду с атрофированными мышечными группами неатрофированных волокон. При электр о миографии выявляется уменьшение амплитуды и длительности потенциала, изменение формы потенциала (полифазность типа группирования или типа потенциалов с короткими пиками). При поражении передних рогов спинного мозга в атрофированных мышцах проксимальных отделов конечностей и туловища выявляются фибриллярные подергивания, асимметрия поражения; рано появляется атрофия и реакция перерождения мышц при исследовании электровозбудимости. При поражении двигательных корешков или волокон в составе периферического нерва возникают парезы или параличи преимущественно в дистальных отделах конечностей, расстройства чувствительности по полиневритическому типу, фибриллярные подергивания отсутствуют. Среди наследственных амиотрофий, протекающих с преимущественным поражением клеток передних рогов спинного мозга, различают болезнь Верднига — Гоффманна, болезнь Кугельберга — Веландера, болезнь Арана— Дюшенна и заболевания с преимущественным поражением периферических нервов (невральная амиотрофия Шарко — Мари — Тута, гипертрофический неврит Дежерина — Сотта). Спинальная амиотрофия Верднига — ГоффманнаСпинальная амиотрофия Верднига — Гоффманна — заболевание, характеризующееся преимущественным перерождением клеток передних рогов спинного мозга. Впервые описано Верднигом (G. Werdnig, 1891) и Гоффманном (J. Hoffmann, 1893) у детей раннего возраста и рассматривалось как типичная форма амиотрофии. Современные представления о заболевании во многом отличаются от классического описания Верднига и Гоффманна. Это относится прежде всего к изменению представлений о времени развития клинических симптомов заболевания, о возможном доброкачественном течении, распространении морфологических изменений за пределы передних рогов спинного мозга. Болезнь нередко имеет семейный характер и передается по аутосомно-рецессивному типу. При патологоанатомическом исследовании выявляются дегенеративные изменения и гибель клеток передних рогов, вторичное перерождение передних корешков и двигательных волокон периферических нервов. В некоторых случаях поражаются клетки задних и боковых рогов грудного отдела спинного мозга, задние и боковые столбы, ядра мозжечка. Клинические проявления весьма разнообразны по тяжести и течению. Наряду с тяжелыми формами болезни, протекающими с развитием мышечной слабости и расстройствами дыхания вскоре после рождения, с нарушениями, приводящими к смерти в первые годы жизни, имеются относительно доброкачественные формы, при которых больные доживают до юношеского или зрелого возраста. Различают раннюю детскую (врожденную), а также детскую и поздние формы заболевания. |
Ранняя детская форма амиотрофии клинически может проявляться во внутриутробном периоде или на первом году жизни. Отмечается позднее и вялое шевеление плода. Ребенок рождается с двигательными нарушениями, почти полным отсутствием движений в первые месяцы жизни. При обследовании обнаруживается отсутствие или снижение сухожильных рефлексов, атрофия мышц, разболтанность суставов. Врожденная миатония описана Оппенгеймом (Н. Oppenheim, 1900). По мнению автора, считавшего ее самостоятельной болезнью, в отличие от амиотрофии Верднига — Гоффманна, при этой форме не отмечается прогрессирования. Ведущим симптомом является атония мышц, мышечная слабость в проксимальных отделах конечностей и туловища, сухожильные рефлексы снижены. Однако многие современные авторы полагают, что врожденная миатония является доброкачественным вариантом ранней формы амиотрофии Верднига — Гоффманна. При ранней детской форме продолжительность жизни от 1 до 7 лет.
Детская форма амиотрофии начинается в возрасте до 4 лет, отличается от первой более медленным течением. Болезнь носит прогрессирующий характер. Длительность заболевания различна. Летальный исход наступает обычно до 14 лет. Поздние формы спинальной амиотрофии: ювенильная форма амиотрофии и форма с более поздним началом — амиотрофия Кугельберга — Веландера. Описаны Кугельбергом и Веландером (Е. Kugelberg, L. Welander, 1954).
Заболевание характеризуется медленно прогрессирующей мышечной слабостью, атрофией мышц, наличием фасцикуляций, отсутствием пирамидных симптомов. Передается по аутосомно-рецессивному типу. Встречается у мужчин в 2 раза чаще, чем у женщин. У большинства больных наблюдается слабость проксимальных отделов верхних и нижних конечностей. Атрофия мышц, возникающая во всех случаях, может быть завуалирована наличием жировой клетчатки, часто обнаруживается гипертрофия ягодичных и икроножных мышц. Заболевание медленно прогрессирует, больные живут в среднем до 40 лет, иногда дольше. В более поздних стадиях в патологический процесс вовлекается мускулатура дистальных отделов конечностей, однако течение заболевания доброкачественное и в поздних его стадиях двигательные функции относительно сохранены, больные способны передвигаться, обслуживать себя.
Течение.
Несмотря на различные варианты течения заболевания, развитие спинальной амиотрофии проходит определенные стадии.
Первая — препаралитическая — стадия характеризуется мышечной гипотонией, слабостью в проксимальных отделах конечностей, снижением рефлексов. Больные свободно передвигаются, мышечная слабость выявляется при физических нагрузках. Во второй стадии — паретической — развиваются глубокие парезы мышц конечностей, снижение рефлексов, слабость дыхательной мускулатуры. Заметно ограничена двигательная активность. На электро-миограмме выявляется спонтанная активность, ритм частокола. Третья стадия — паралитическая. Больные обездвижены, у них отмечается излишнее отложение жира на конечностях и туловище, что часто маскирует атрофию мышц и фибриллярные подергивания. Параличи выражены в проксимальных и дистальных отделах конечностей. Четвертая стадия — кахектическая — характеризуется общим похуданием и диффузной атрофией мышц, спонтанными фасцикуляциями в мышцах. На электромиограмме — характерный ритм частокола. Пятая стадия — терминальная — характеризуется развитием контрактур с грубой деформацией скелета, при этом мелкие суставы кистей и пальцев стоп остаются менее пораженными.
При всех вариантах для спинальной амиотрофии характерно развитие парезов, параличей преимущественно проксимальных отделов конечностей. В большей степени поражаются сгибатели нижних конечностей. Характерно отсутствие глубоких рефлексов, фасцикулярные и фибриллярные подергивания скелетных мышц и мышц языка. Вследствие поражения дыхательных мышц возникает спинальный тип расстройства дыхания (ограничение подвижности грудной клетки, участие в акте дыхания вспомогательных мышц, снижение объема легочной вентиляции, артериальная гипоксемия и гиперкапния). Чувствительность и координационная сфера не нарушены, функция тазовых органов сохранена, интеллект не изменен.
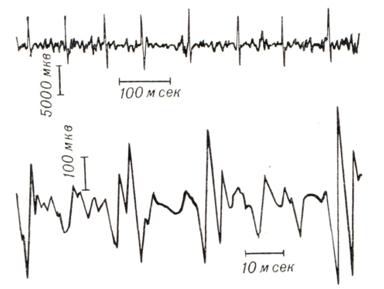
Электромиографическое исследование подтверждает неврогенный характер повреждения (ритм частокола, полифазия, фибрилляции и фасцикуляций (рис. 1).
 |
 |
 |
Рис. 1. | ||
Прогноз в тяжелых случаях неблагоприятный в связи с отсутствием специфической терапии. Непосредственной причиной смерти является пневмония или интеркуррентные инфекции.
Спинальная амиотрофия Арана — Дюшенна.
Заболевание впервые было описано Дюшенном (G. В. A. Duchennе) в 1849 г. и Араном (F.A. Aran) в 1850 г. Нозологическая самостоятельность заболевания оспаривается многими авторами. Однако С. Н. Давиденков подчеркивает, что спинальная амиотрофия взрослых существует в качестве самостоятельной формы. В основе заболевания лежит хронический дегенеративный процесс в клетках передних рогов спинного мозга, передних корешках и периферических нервах. При гистологическом исследовании мышечных волокон выявляется пучковый характер атрофии, при электромиографическом — изменения, характерные для невральных атрофии.
Заболевание начинается в возрасте 40—60 лет. Характерной клинической особенностью заболевания является медленно прогрессирующая мышечная атрофия в начальных стадиях, преимущественно в дистальных отделах конечностей («обезьянья кисть»). Атрофии, как правило, симметричны. По мере развития заболевания в процесс вовлекаются мышцы плечевого пояса, проксимальных отделов конечностей, реже мышцы глотки, гортани, языка и другие.
Заболевание медленно, в течение ряда лет, прогрессирует. Летальный исход наступает обычно в результате интеркуррентных заболеваний.
Невральная амиотрофия Шарко — Мари — Тута
Невральная амиотрофия Шарко — Мари — Тута — заболевание, относящееся к группе невральных А. Описано Шарко и Мари (J. M. Charcot, P. Marie) в 1886 г. во Франции и Тутом (Н. Н. Tooth) в 1886 г. в Англии. Заболевание начинается в различном возрасте (преимущественно в возрасте 10—30 лет), чаще наблюдается у лиц женского пола. Передается по аутосомно-доминантному, реже — по аутосомно-рецессивному типу и рецессивному, сцепленному с полом.
Заболевание характеризуется атрофией дистальных отделов конечностей. Атрофия начинается обычно с мышц голени, стопы, преимущественно малоберцовых («перо-неальная» стопа, «перонеальная» походка). Через нек-рое время возникает слабость и атрофия дистальных отделов верхних конечностей. Сухожильные рефлексы снижаются или угасают полностью. Наряду с двигательными нарушениями выявляются дистальный тип расстройства чувствительности, трофические расстройства (цианоз, отек, краснота, гипергидроз, ангидроз). Заболевание медленно прогрессирует. Стопы деформируются по типу стопы Фридрейха. Атрофия мышц кисти приводит к деформации руки по типу обезьяньей кисти или «выеденной ладони». Мышцы проксимальных отделов, как правило, сохранены, что дает возможность больным длительное время самостоятельно передвигаться, обслуживать себя и сохранять трудоспособность.
Существует ряд атипичных, переходных форм заболевания, что затрудняет дифференциальную диагностику. В основе заболевания лежит так называемая хронеская периферическая невропатия (поражение нервов и изменение в интерстициальной соединительной ткани), изменения в двигательных клетках спинного мозга и в некоторых случаях в боковых и задних столбах спинного мозга. В мышцах в начальный период заболевания отмечается пучковый характер атрофии, в дальнейшем развивается гиперплазия соединительной ткани, выявляются дистрофические изменения мышечных волокон.
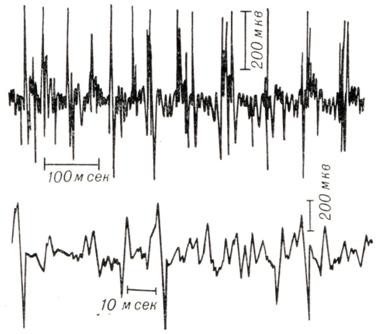
Электромиографическое исследование позволяет обнаружить патологические изменения на ранних стадиях заболевания; при этом выявляются фибрилляции, длинные денервационные потенциалы, синхронизация потенциалов (рис. 2).
|
 |
|
Рис. 2. | ||
Гипертрофический неврит (семейная гипертрофическая невропатия) Дежерина — Сотта.
Нозологическая самостоятельность этого заболевания не доказана. Одни авторы рассматривают гипертрофический неврит как один из вариантов невральной амиотрофии, другие — как самостоятельную форму болезни (см. Дежерина — Сотта гипертрофический неврит).
Диагноз ставят на основании клинической картины и данных лабораторных исследований.
Биохимические исследования. При амиотрофии установлено умеренное повышение активности ферментов: альдолазы, креатинфосфокиназы, траысаминаз в сыворотке крови. Однако по сравнению с первичными миопатиями это повышение выражено значительно слабее. Другим характерным биохимическим признаком амиотрофии является креатинурия, отражающая степень атрофии мышечной ткани одновременно у больных снижается экскреция с мочой креатинина. Креатининовый показатель мочи, представляющий отношение креатинина к сумме креатина и креатинина и в норме равный единице, снижается до 0,8 (С. Н. Давиденков). Содержание в плазме крови и в моче у больных амиотрофией свободных аминокислот существенно не меняется. Наследственные амиотрофии следует дифференцировать с мышечными атрофиями (смотри Атрофия мышечная). Прогноз неблагоприятный при ранних и быстротекущих формах амиотрофи и — больные погибают в детском возрасте. При поздних и медленно текущих формах прогноз относительно благоприятный: больные благодаря сохранным мышечным группам могут компенсировать функцию пораженных мышц. Следует избегать переохлаждения и переутомления, ведущих к обострению заболевания.
Родителям, имеющим ребенка, больного спинальной или невральной амиотрофией, рекомендуется воздержание от дальнейшего деторождения.
Лечение. При всех формах амиотрофий рекомендуется АТФ, витаминотерапия (витамины группы В, витамин Е), аминокислоты (метионин, глутаминовая кислота и другие), анаболические гормоны (неробол, ретаболил), аитихолинэстеразные препараты (прозерин, галантамин, нивалин). Лечение проводится курсами, длительность каждого курса составляет 1 —1½ месяца. В год проводится несколько курсов лечения. Особое внимание должно быть направлено на предотвращение контрактур (массаж, гимнастика).
|
Бадалян Л.О. |

|
⇐ Перейти на главную страницу сайта |
⇑ Вернуться в начало страницы ⇑ |
Библиотека Ordo Deus ⇒ |
⇐ Амиотрофии |
⇓ Каталог систематический ⇓ |
Амиотрофический боковой склероз (БМЭ) ⇒ |
|
Все статьи в полном изложении, Вы можете найти в большой медицинской энциклопедии — Главный редактор: академик АН СССР (РАН) и АМН СССР (РАМН) Б.В. Петровский. — Москва издательство «Советская энциклопедия» 1989г. |
|
Внимание! Вы находитесь в библиотеке «Ordo Deus». Все книги в электронном варианте, содержащиеся в библиотеке «Ordo Deus», принадлежат их законным владельцам (авторам, переводчикам, издательствам). Все книги и статьи взяты из открытых источников и размещаются здесь только для чтения. |
|
Библиотека «Ordo Deus» не преследует никакой коммерческой выгоды. |
|
Все авторские права сохраняются за правообладателями. Если Вы являетесь автором данного документа и хотите дополнить его или изменить, уточнить реквизиты автора, опубликовать другие документы или возможно вы не желаете, чтобы какой-то из ваших материалов находился в библиотеке, пожалуйста, свяжитесь с нами по e-mail: |
Вас категорически не устраивает перспектива безвозвратно исчезнуть из этого мира? Вы желаете прожить ещё одну жизнь, начав всё заново? Вы желаете прожить ещё одну жизнь без ошибок совершённых в этой жизни? Вы желаете прожить ещё одну жизнь, осуществив в ней мечты несбывшиеся в этой жизни? Перейдите по ссылке: «главная страница».
|
© Ordo Deus, 2010. При копировании ссылка на сайт http://www.ordodeus.ru обязательна. |