Гаргоилизм |
||
 |
 |
|
Оглавление
|
ГаргоилизмГаргоилизм (французский gargouille рыльце водосточной трубы в соборах готической архитектуры, украшавшееся фантастическими фигурами с уродливыми лицами; синонимы: множественный дизостоз, липохондродистрофия, полидистрофия, остеохондродистрофия) — заболевание, обусловленное тяжёлой наследственной патологией соединительной ткани, проявляющееся сочетанным поражением костно-суставной системы, глаз, внутренних органов и нервной системы. Впервые описано Гунтером (С. Hunter, 1917) и Гурлер (G. Hurler, 1919). Эллис (R. Ellis, 1936) предложил термин «гаргоилизм» из-за характерного внешнего вида больных. К Гаргоилизм относят первые два типа мукополисахаридозов (смотри) — синдром Гурлер и синдром Гунтера. Джервис (G. A. Jervis, 1950) установил, что синдром Гурлер наблюдается чаще других мукополисахаридозов (1 : 10 000 рождений). По данным ВОЗ (1968), синдром Гунтера встречается в 1 случае на 200 000 рождений. Этиология и патогенезДля синдрома Гурлер характерен аутосомнорецессивный тип наследования, при синдроме Гунтера — наследование рецессивное, сцепленное с полом. Установлено, что в основе Гаргоилизм лежат генетически обусловленные нарушения обмена кислых мукополисахаридов (смотри), связанные с нарушением структуры, избыточным накоплением их в тканях организма и повышенным выведением с мочой. Определённое значение в патогенезе мукополисахаридозов отводится ферментам, участвующим в расщеплении кислых мукополисахаридов. О’Брайен (D. O’Brien) с сотрудники (1969) и Оккерман (P. A. Ockerman, 1969) сообщили о дефиците β-галактозидазы в печени и повышении её активности в плазме и моче больных. Выделенные Дорфманом и Лоринцем (A. Dorfman, A. Lorincz, 1957) и Мейером (К. Meyer, 1958) из мочи больных при синдроме Гурлер кислые мукополисахариды оказались смесью гепарансульфата и дерматансульфата, общее количество их достигало 150 миллиграмм и более в сутки. В норме экскреция кислых мукополисахаридов с мочой не превышает 10 в 1 суток. Ряд авторов указывает на преобладание при синдроме Гурлер дерматансульфата, другие — гепарансульфата. В. Мак-Кьюзик (1965) и М. Р. Гусева (1972) считают, что при Гаргоилизм эти мукополисахариды могут выделяться в разных соотношениях, при этом преобладает и усиливается с возрастом экскреция дерматансульфата, обнаруживается высокая экскреция с мочой хондроитинсульфатов. У больных Гаргоилизм выявлены значительные изменения метаболизма липидов, преимущественно гликолипидов (сульфатидов), холестерола и отдельных фракций фосфолипидов. Наиболее резкие изменения отмечены при синдроме Гурлер. Характерным также является нарушение обмена коллагена, что проявляется низким содержанием оксипролина (смотри Пролин) в моче (в 2—3 раза ниже нормы). Имеются нарушения в обмене других аминокислот — избыточное содержание в моче серина и треонина, участвующих в связях полипептидов и мукополисахаридов. Патологическая анатомияПри морфологически исследовании выявляется поражение различных органов и систем с накоплением высокомолекулярных веществ липоидно-полисахаридной природы. Эти вещества откладываются в виде мелкозернистой массы в клетках мозга, сетчатки глаз, периферических нервных ганглиев. Нервные клетки представляются набухшими, увеличенными в размерах, ядро сдвинуто к периферии. Субстанция Ниссля исчезает или уменьшается, видны лишь остатки, сдвинутые в сторону вместе с ядром. Твёрдая мозговая оболочка обычно утолщена. Гомогенные зернистые массы откладываются также в клетках опорной ткани, роговицы, внутренних органов (печени, селезёнки, эпителия дистальных отделов извитых канальцев почек). В клетках печени и почек наряду с кислыми мукополисахаридами откладывается значительное количество нейтральных жиров. Характерным является изменение костей — нарушение энхондрального окостенения, разрастание кровеносных сосудов в хряще и накопление в его клетках кислых мукополисахаридов и липидов. |
В межуточном веществе соединительной ткани выявляются только растворимые мукополисахариды, липоидные вещества не обнаруживаются. Морфологически выявляется дезорганизация структуры основного вещества, утолщение и гомогенизация коллагеновых волокон. В значительной степени страдают мозг, печень, селезёнка, глаза. Наблюдаются патологический изменения в сердечных клапанах, аорте, кровеносных сосудах мозга.
Клиническая картина
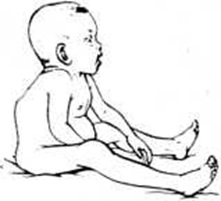
При синдроме Гурлер больной имеет характерный внешний вид. Уже на первом году жизни развиваются типичные для данного заболевания изменения конфигурации черепа, деформация позвоночника (рисунок 1). Эти изменения часто расцениваются как признаки рахита. К одному году состояние больных ухудшается, поясничный кифоз и тугоподвижность суставов резко усиливаются. Дети отстают в физ. и умственном развитии. После одного года клин, симптомы становятся особенно выраженными. Для больных Гаргоилизм характерны: запавшая переносица, нависший лоб, толстые губы и язык, широко расставленные зубы, деформация и увеличение в размерах черепа, отставание в росте, нижнегрудной и поясничный кифоз, деформация грудной клетки, тугоподвижность суставов, пупочные и паховые грыжи, гепато- и спленомегалия, изменения со стороны сердца (систолический шум, приглушение тонов, расширение границ сердца).
 |
 |
 |
Рис. 1. | ||
На ЭКГ выявляется диффузное поражение миокарда. Поражение органа зрения наблюдается у всех больных и является наиболее ранним и типичным симптомом заболевания.
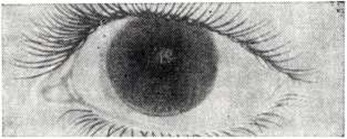
У большинства больных развивается помутнение роговицы, которое возникает в глубоких слоях, а затем диффузно захватывает всю роговицу (рисунок 2). В отдельных случаях помутнение роговицы может быть выявлено с рождения. Нередко наблюдаются мегалокорнеа, врождённая глаукома, застойные явления и атрофия дисков зрительного нерва, пигментная дистрофия сетчатки.
|
 |
|
Рис. 2. | ||
Значительные изменения наблюдаются со стороны нервной системы; резко выраженный гипертензионно-гидроцефальный синдром, изменения мышечного тонуса и сухожильных рефлексов, иногда выявляются пирамидные знаки, в отдельных случаях — поражение черепных нервов (косоглазие, нистагм), вегетативно-трофические расстройства (избыточное питание, гипертрихоз), эндокринные изменения (микседема).
Психические расстройства проявляются слабоумием, которое достигает обычно выраженной степени (вплоть до идиотии), сопровождается нарушениями речи и иногда снижением слуха. У больных наблюдается повышенная истощаемость нервной системы, иногда возникают эпилептиформные припадки.
Большинство исследователей, исходя из того, что при Гаргоилизм происходит прогрессирующее накопление в клетках различных органов, в том числе головного мозга, патологический продуктов нарушенного обмена, рассматривает слабоумие при Гаргоилизм как результат прогредиентного процесса и относит его к приобретённому слабоумию (смотри). В пользу такой точки зрения свидетельствует прежде всего течение заболевания, первые признаки которого появляются после рождения и в дальнейшем неуклонно нарастают, в том числе слабоумие. В отличие от олигофрении, слабоумие при Гаргоилизм характеризуется мозаичностью. В частности, отмечается несоответствие между степенью снижения интеллекта и нарушением деятельности, которая в силу слабости побуждений расстроена относительно больше. Преимущественные расстройства механической (а не ассоциативной) памяти, астенические и эпилептиформные проявления также больше соответствуют органической деменции, чем истинной олигофрении.
Синдром Гунтера протекает более благоприятно. Клинической, симптомы заболевания проявляются обычно в 3—4 года. До 1 года дети развиваются нормально. Характерны ранние изменения скелета в виде увеличения размеров черепа. Тугоподвижность и изменения формы суставов становятся выраженными к 3 годам, изменения скелета в виде деформации грудной клетки и позвоночника умеренные.
В росте больные отстают в меньшей степени, чем при синдроме Гурлер. Черты лица менее грубые. Гепатоспленомегалия и поясничный кифоз у ряда больных отсутствуют. Могут выявляться пороки сердца. Характерно наличие пупочной и паховой грыжи, снижение слуха. Помутнение роговицы отсутствует или выражено незначительно, часто выявляется мегалокорнеа, застой и атрофия дисков зрительных нервов, пигментная дистрофия сетчатки. Снижение интеллекта выражено в меньшей степени, чем при синдроме Гурлер.
Диагноз основывается на клинической, рентгенологическое и лабораторных данных. Большое диагностическое значение имеет биохимический исследование мочи больных на кислые мукополисахариды: реакция Берри с толуидиновым синим, реакция преципитации с цетавлоном, с альбумином (смотри Экспресс-методы). Отдельные фракции кислых мукополисахаридов могут быть изучены с помощью ионообменной хроматографии и электрофореза на бумаге или ацетатцеллюлозе.
При Гаргоилизм выделяются в больших количествах дерматансульфат, гепарансульфат, а также хондроитинсульфаты. С возрастом экскреция с мочой дерматансульфата увеличивается.
Диагностическую ценность может иметь обнаружение метахроматических грануляций в лейкоцитах крови и клетках костного мозга (гранулы Рейли).
Наряду с клинической, и биохимический исследованием больных Гаргоилизм необходимо изучение родословной и установление типа наследования, а также исследование экскреции кислых мукополисахаридов в моче родителей и родственников больных, у которых фракционный состав кислых мукополисахаридов может быть изменён.
Для обнаружения гетерозиготного носительства большое значение имеет изучение культуры фибробластов.
Рентгенодиагностика
В типичных случаях Гаргоилизм без труда распознается по рентгенограммам.
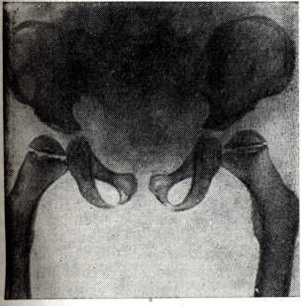
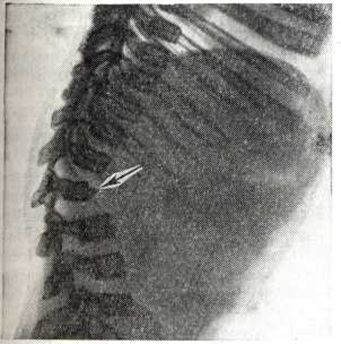
Заболевание характеризуется генерализованным симметричным поражением эпифизов костей. Они уплощены и увеличены. Метадиафизарные отделы длинных и коротких трубчатых костей укорочены и утолщены. Типичной является вальгусная деформация проксимальных отделов бедренных костей (рисунок 3). Плоские кости деформированы, утолщены, суставные впадины уплощены. Ребра также утолщены, передние концы их булавовидно расширены. Шейки рёбер остаются нормальными. Межрёберные промежутки сужены. Позвоночник всегда поражён, имеют место платиспондилия и угловой кифоз в нижнегрудном и верхнепоясничном отделе (рисунок 4). Патогномоничным является недоразвитие передне-верхнего угла тел позвонков, обычно ThXII или LI-II. Межпозвонковые пространства не сужены. Имеется диспропорция в развитии лицевого и мозгового черепа. Основание черепа укорочено. Турецкое седло уплощено, удлинено, передние клиновидные отростки недоразвиты. Кости лицевой части черепа, особенно верхняя челюсть и носовые кости, недоразвиты. Задерживается пневматизация придаточных пазух носа. Изменяются темпы и последовательность окостенения, остеогенез происходит асимметрично.
|
 |
|
Рис. 3. | ||
|
 |
|
Рис. 4. | ||
Дифференциальный диагноз на ранних стадиях заболевания необходимо проводить с врождённой гидроцефалией (смотри), рахитом (смотри), гипотиреозом (смотри), липоидозами (смотри). Дифференциальная диагностика проводится также с другими формами хондродистрофии (смотри) и эпифизарной дисплазии. При них, в отличие от Гаргоилизм, отмечается нормальная толщина диафизов трубчатых костей. Эпифизы имеют неправильную форму, увеличены, очертания их неровные, структура фрагментирована. Типична изодактилия. Таз деформирован, вдавлен с боков, вертлужные впадины расположены глубже, уплощены. В отличие от Гаргоилизм наблюдается варусная деформация бедренных костей, определяется системная брахиспондилия и ангуляция позвонков. При этом патогномоничной является деформация одного или двух позвонков в виде конусовидного, клиновидного заострения переднего отдела тел в нижнегрудном или верхнепоясничном отделах.
Лечение
Целесообразно назначение гормональных препаратов, в частности глюкокортикоидных гормонов и АКТГ, препятствующих синтезу кислых мукополисахаридов. Эффективно применение тиреоидина в связи с отмечающейся у больных гипофункцией щитовидной железы.
Некоторые авторы предлагали применять тиреоидин в сочетании с облучением гипофиза. Возможно улучшение при сочетанном применении) тиреоидина и преднизолона.
Предприняты попытки лечения мукополисахаридозов большими дозами витамина А. Кроме того, применяют стимулирующие средства: глутаминовую кислоту, церебролизин, аминалон (гаммалон), витамины группы В. При наличии судорожных припадков назначают противосудорожные средства, при астении — общеукрепляющую терапию. Учитывая пониженную сопротивляемость организма больных Гаргоилизм, следует оберегать их от инфекционных и других интеркуррентных заболеваний.
Прогноз
Болезнь неуклонно прогрессирует. Летальный исход наступает от заболеваний дыхательных путей и сердечной декомпенсации. Наиболее тяжёлым является первый тип мукополисахаридоза (синдром Гурлер), характеризующийся прогрессирующим течением и ранней смертью больных (большинство погибает до 10—12-летнего возраста). Мукополисахаридоз второго типа (синдром Гунтера) протекает менее злокачественно — больные доживают до 30 и более лет. При проведении ранней терапии прогноз более благоприятный.
Профилактика. Родителям, имеющим больного ребёнка, рекомендуется воздержаться от дальнейшего деторождения. Возможна внутриутробная диагностика заболевания на третьем месяце беременности путём исследования культуры клеток амниотической жидкости, полученной при амниоцентезе (смотри).
|
Вельтищев Ю.E.; Врно М.С.; Гусев E.И.; Гусева М.P.; Климова М.К. |

|
⇐ Перейти на главную страницу сайта |
⇑ Вернуться в начало страницы ⇑ |
Библиотека Ordo Deus ⇒ |
⇐ Ганзера синдром |
⇓ Полный свод знаний. Том первый А. ⇓ |
Гарднера синдром ⇒ |
|
Все статьи в полном изложении, Вы можете найти в большой медицинской энциклопедии — Главный редактор: академик АН СССР (РАН) и АМН СССР (РАМН) Б.В. Петровский. — Москва издательство «Советская энциклопедия» 1989г. |
|
Внимание! Вы находитесь в библиотеке «Ordo Deus». Все книги в электронном варианте, содержащиеся в библиотеке «Ordo Deus», принадлежат их законным владельцам (авторам, переводчикам, издательствам). Все книги и статьи взяты из открытых источников и размещаются здесь только для чтения. |
|
Библиотека «Ordo Deus» не преследует никакой коммерческой выгоды. |
|
Все авторские права сохраняются за правообладателями. Если Вы являетесь автором данного документа и хотите дополнить его или изменить, уточнить реквизиты автора, опубликовать другие документы или возможно вы не желаете, чтобы какой-то из ваших материалов находился в библиотеке, пожалуйста, свяжитесь с нами по e-mail: |
Вас категорически не устраивает перспектива безвозвратно исчезнуть из этого мира? Вы не желаете закончить свой жизненный путь в виде омерзительной гниющей органической массы пожираемой копошащимися в ней могильными червями? Вы желаете вернувшись в молодость прожить ещё одну жизнь? Начать всё заново? Исправить совершённые ошибки? Осуществить несбывшиеся мечты? Перейдите по ссылке: «главная страница».
|
© Ordo Deus, 2010. При копировании ссылка на сайт http://www.ordodeus.ru обязательна. |