Гликогеноз |
||
 |
 |
|
Оглавление
|
ГликогенозГликогеноз (glycogenosis, единственное число; гликоген + -osis; синонимы: гликогенная болезнь, гликогенная аккумуляция) — группа наследственных энзимопатий, возникающих в связи с дефицитом ферментов, катализирующих процессы распада или синтеза гликогена, и характеризующихся избыточным его накоплением в различных органах и тканях. Впервые больной гликогенозом был описан в 1910 год Леревуайлет (Lerevouillet). В 1928 год Ван-Кревельд (S. van Creveld) описал клинический, картину Гликогеноз I типа, а в 1929 год Гирке (Е. van Gierke) патологоанатомическую картину этого заболевания, установив при нем накопление гликогена в печени и почках. Первое энзимологический исследование Гликогеноз выполнено в 1952 год Помпе (I. С. Pompe).
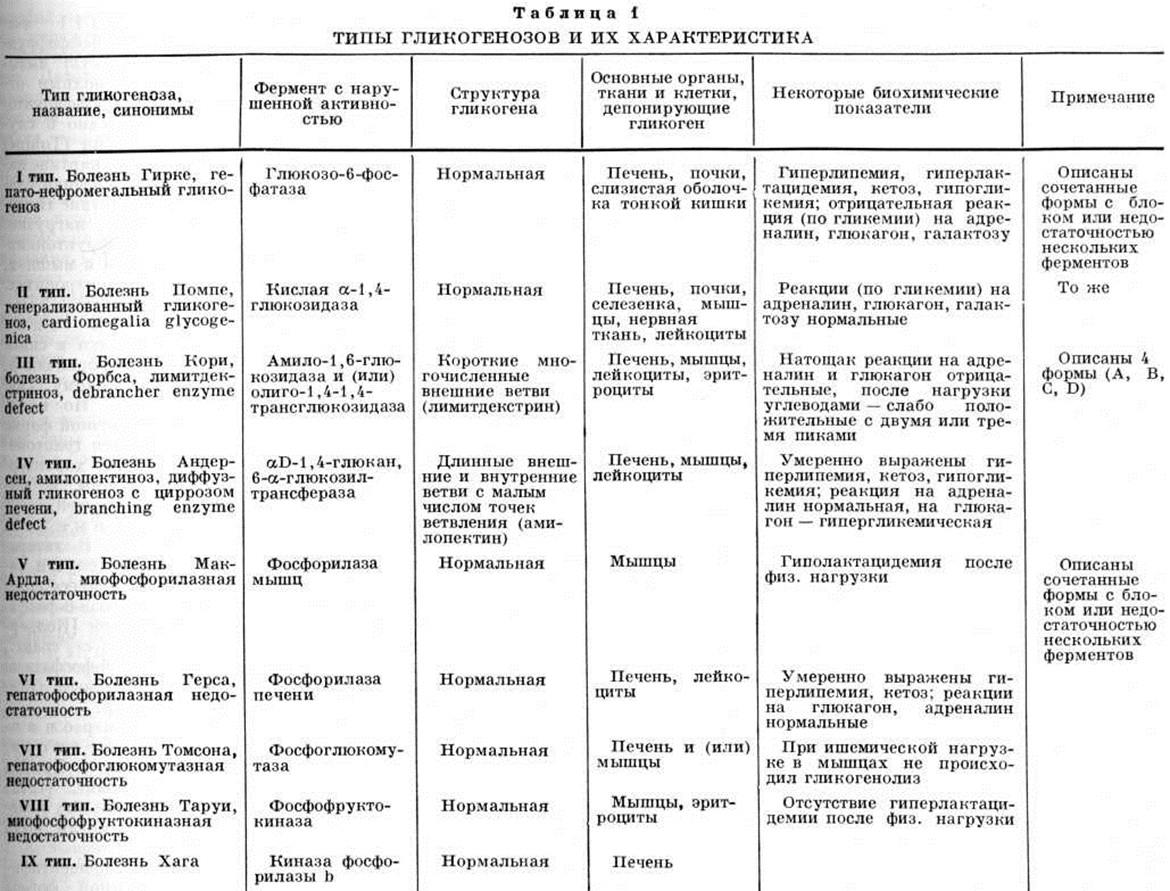
Распространённость Гликогеноз в популяции составляет 1 : 40 000. Выделено 12 типов Гликогеноз, наиболее полно изучены 9 (таблица 1); типы X (изолированный Гликогеноз сердца) и XI (дефицит фосфофруктокиназы), возможно, являются вариантами основных форм Гликогеноз. Типы гликогенозовРазличают три основные формы Гликогеноз: печёночную, мышечную и генерализованную. Гликогеноз I типа (болезнь Гирке, гепатонефромегальный Гликогеноз) связан с дефицитом активности глюкозо-6-фосфатазы печени и почек, что может быть установлено при жизни с помощью гистохимические исследования материала, полученного при биопсии этих органов. Наследуется по аутосомно-рецессивному типу. По клинической картине это заболевание относят к печёночной форме Гликогеноз. Первые проявления его — отсутствие аппетита, рвота, гипогликемические судороги (комы), респираторный дистресс-синдром (смотри полный свод знаний), интермиттирующее повышение температуры, гепатомегалия, нефромегалия, стеаторея (смотри полный свод знаний), кетонурия (смотри полный свод знаний Ацетонурия) — выявляются сразу же после рождения или в грудном возрасте. |
|||||||||||||
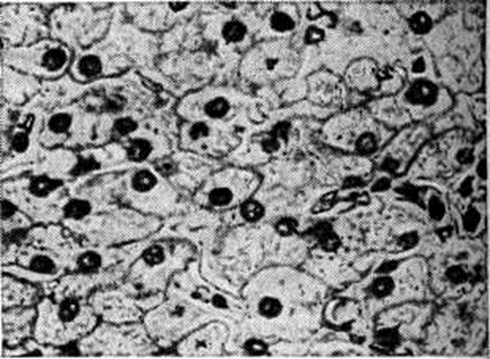
С возрастом прогрессируют гепато и нефромегалия за счёт гликогенной инфильтрации (рисунок 1), отставание в росте, диспропорция тела (большая голова, короткие шея и ноги), кукольное лицо, гипотония мышц; нарастает гипогликемический синдром натощак (больные вынуждены почти постоянно принимать пищу). Часто присоединяющиеся вторичные инфекции резко усиливают кетоацидоз (смотри полный свод знаний Ацидоз) и гипогликемию (смотри полный свод знаний) и нередко являются причиной смерти. Реже наблюдаются геморрагический синдром, кожный ксантоматоз. Нервнопсихическое развитие удовлетворительное; половое созревание значительно задерживается. Состояние больных несколько улучшается в пубертатном периоде.
 |
|  |
Рис. 2. | ||
Биохимический нарушения: гипогликемия, кетоз, гиперлактацидемия, гиперлипемия, повышение в крови уровня неэстерифицированных жирных кислот, гликогена, холестерина, мочевой кислоты, нарушение почечного клиренса для ряда веществ. Обнаруживается интолерантность к глюкозе. Введение адреналина, глюкагона, галактозы вызывает значительную гиперлактацидемию, но не гипергликемию, так как глюкозо-6-фосфатаза в печени отсутствует.
Окончательный диагноз ставится в результате исследования активности глюкозо-6-фосфатазы в печени путём прижизненной биопсии. Ван-Хоф (F. van Hoof) с соавторами в 1972 год предложил для диагностики I типа Гликогеноз использовать меченную по 14С и 3Н глюкозу.
|
| |
| ||
Прогноз определяется степенью снижения активности фермента. Дети погибают от ацидотической комы или от интеркуррентных заболеваний.
Гликогеноз II типа (болезнь Помпе, генерализованный гликогеноз, cardiomegalia glycogenica) развивается в связи с дефицитом кислой α-1,4-глюкозидазы. Наследование аутосомно-рецессивное, отмечается связь с частотой возникновения эндокардиального фиброэластоза [Динской (М. Y. Dinscoy) с соавт., 1965].
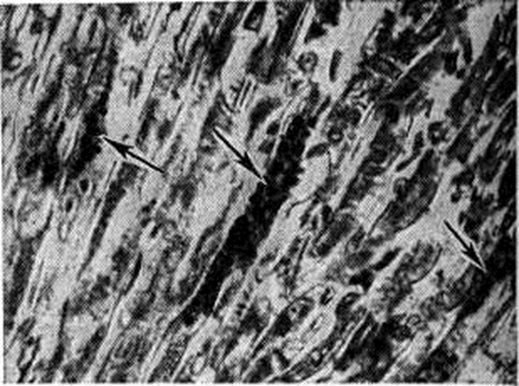
При патологоанатомическом исследовании обнаруживаются отложения гликогена во всех органах и тканях. Размеры сердца увеличены за счёт гликогенной инфильтрации мышечных волокон (рисунок 2). Органических изменений в клапанном аппарате сердца не отмечается. Отложение гликогена в мышечных волокнах языка нередко ведёт к макроглоссии, в мышечных волокнах пилорического отдела желудка — к пилороспазму, в диафрагме и других дыхательных мышцах — к дыхательной недостаточности. Печень, селезёнка и почки равномерно увеличены. По клинический, картине это заболевание относят к генерализованной форме Гликогеноз Первые симптомы выявляются через несколько дней или недель (до 6 мес.) после рождения: цианоз (общий и интермиттирующий), расстройство дыхания (ускоренное, поверхностное), беспокойство, апатия и адинамия. Позже присоединяются макроглоссия, миогипотония. Рано развивается гепатомегалия, пилороспазм. В дальнейшем ведущими в клинический, картине становятся кардиомегалия (шаровидное сердце, изменения ЭКГ), одышка, бронхиты, ателектазы, гипостатические пневмонии, миодистрофия, гипорефлексия, бульбарные нарушения, спастические параличи. Отмечается отсутствие аппетита, задержка роста.
В сыворотке крови повышено содержание мочевой кислоты, глутаминщавелево-уксусной трансаминазы и альдолазы; в мышцах, печени, лейкоцитах — дефицит кислой (лизосомальной) α-1,4-глюкозидазы, содержание гликогена в печени — 12%, в мышцах— 10%. Рано появляется генерализованное отложение гликогена.
Наряду с генерализованной формой встречаются случаи заболевания, вызванные дефицитом кислой а-глюкозидазы только в мышцах. В этих случаях болезнь, как правило, проявляется в более позднем возрасте и по клинический, картине напоминает мышечные формы Гликогеноз
Решающим в диагностике является биохимический исследование ткани, полученной при биопсии. Генерализованный характер Гликогеноз II типа позволяет определять активность глюкозидазы в форменных элементах крови и в коже больного, а также в культуре клеток фибробластов кожи и мышц больного.Возможна пренатальная диагностика, основанная на биохимический исследовании клеток слущивающегося эпителия кожи плода, находящихся в амниотической жидкости.
|

| |
| ||
Прогноз при генерализованной форме неблагоприятный, больные погибают на первом году жизни от сердечной или дыхательной недостаточности. При накоплении гликогена только в скелетных мышцах прогноз более благоприятный.
Гликогеноз III типа (болезнь Кори, болезнь Форбса, лемитдекстриноз, debrancher enzyme defect) вызывается полным или частичным отсутствием активности амило-1, 6-глюкозидазы и (или) олиго-1,4-1,4-трансглюкозидазы. Установлено 4 подтипа заболевания (таблица 2). Генетический анализ этого типа Гликогеноз труден из-за наличия нескольких его форм. По клинический, картине относят к мышечной или печеночной форме Гликогеноз
По клинический, картине Гликогеноз этого типа напоминает Гликогеноз I типа: наблюдается гепатомегалия с первых месяцев жизни, мышечная гипотония, гипертрофия отдельных мышечных групп, гипертрофия миокарда, нарушение сердечной проводимости и кровообращения. При биохимический исследованиях отмечается гипогликемия натощак, кетоз, липемия, повышение уровня гликогена в эритроцитах. Расщепление гликогена задерживается (смотри полный свод знаний таблица 1).
Определение активности амило-1,6-глюкозидазы и олиго-1,4-1,4-траисглюкозидазы в мышцах и печени позволяет установить форму Гликогеноз.
Прогноз, как правило, благоприятный. После 5-летнего возраста и особенно в пубертатном периоде развитие заболевания значительно замедляется.
Гликогеноз IV типа (болезнь Андерсена, амилопектиноз, диффузный гликогеноз с циррозом печени, branching enzyme defect) вызывается отсутствием αD-1,4-глюкан, 6-α-глюкозилтрансферазы. Предполагается аутосомно-рецессивный или связанный с полом тип наследования.
Болезнь проявляется с первых лет жизни и характеризуется гепатоспленомегалией, развитием цирроза печени, желтухой, гипогликемией.
Прогноз плохой. Больные обычно погибают на первом году жизни.
Гликогеноз V типа (болезнь Мак-Ардла, миофосфорилазная недостаточность) развивается в связи с дефицитом мышечной фосфорилазы. Активность печёночной фосфорилазы не изменена. Аутосомно-рецессивный тип наследования. Лица мужского пола болеют в 5 раз чаще. В связи с гликогенной инфильтрацией скелетные мышцы увеличиваются в объёме, становятся резко плотными. По клинический, картине заболевание относят к мышечной форме Гликогеноз Симптомы его — мышечная слабость, мышечные спазмы, тахикардия — появляются в первые десять дней жизни и прогрессируют. Появляется транзиторная миоглобинурия [Мак-Ардл (В. Mac Ardle), 1951; Пирсон (С. М. Pearson) с сотрудники, 1961]. Концентрация лактата в крови уменьшается после физических нагрузки. Окончательный диагноз возможен при исследовании фосфорилазной активности в мышечных биоптатах; в гомогенатах мышц гликоген не превращается в лактат.
Прогноз для жизни благоприятный, выздоровление невозможно.
Гликогеноз VI типа (болезнь Герса, гепатофосфорилазная недостаточность) вызывается недостаточностью фосфорилазы в печени. Предполагается аутосомно-рецессивный тип наследования. По клинический, картине относят к печёночной форме Гликогеноз Характерны значительная гепатомегалия в результате гликогенной инфильтрации гепатоцитов, задержка роста, кукольное лицо, гиперлипемия, гипергликемия (после внутривенного введения галактозы), повышенное содержание гликогена в эритроцитах. Прогноз сомнителен.
Гликогеноз VII типа (болезнь Томсона) развивается в связи с дефицитом фосфоглюкомутазы в печени и (или) мышцах. Впервые описан в 1963 год Томсоном (W. Н. S. Thomson) с соавторами у мальчика с миопатией. В 1964 год Иллингворт (В. Illingworth) и Браун (D. Н. Brown) описали мальчика, больного Гликогеноз с гепатомегалией. Встречается редко.
Гликогеноз VIII тип а (болезнь Таруи, миофосфофруктокиназная недостаточность) вызывается дефицитом или полным отсутствием активности фосфофруктокиназы в мышцах. Описано 6 случаев этого типа Гликогеноз [Тобин (Tobine) с соавторами, 1973]. По клинический, картине напоминает Гликогеноз V типа: мышечная слабость, утомление и отсутствие гиперлактацидемии после физ. нагрузки. Низкий уровень фосфофруктокиназы у больных обнаружен в мышцах, у их родителей — в эритроцитах. Прогноз благоприятный.
Гликогеноз IX типа (болезнь Хага) развивается в связи с дефицитом киназы фосфорилазы b. Наследуется по рецессивному, связанному с полом типу. По клинический, картине относят к печёночной форме Гликогеноз. У больных наблюдается гепатомегалия. Другие симптомы, характерные для печёночной формы, не выражены. Прогноз неизвестен.
Комбинированные типы гликогеноза. В литературе описаны отдельные случаи заболевания, характеризующиеся сочетанием отсутствия глюкозо-6-фосфатазы с лимитдекстринозом [Колдербанк (A. Calderbank) с соавторами, 1960], дефицита глюкозо-6-фосфатазы с branching enzyme [Беркофф (Berkoff) с соавторами, 1962] и другие.
Неидентифицированные типы гликогеноза. По данным Иллингворта и Брауна (1964), Ван-Хофа с соавторами (1972) и другие, около 1/3 случаев больных печёночной формой Гликогеноз не могли быть идентифицированы.
Известны случаи неидентифицированных Гликогеноз мышечной формы (В. М. Казаков и соавторами, 1971, В. С. Лобзин и соавторами, 1973, А. А. Шутов и соавторами, 1974). В 1970 год Л. О. Бадалян с сотрудниками наблюдал 2 случая заболевания, по клинический, картине сходных с Гликогеноз V типа. Однако активность фосфорилазы, амило-1,6-глюкозидазы, фосфоглюкомутазы, кислой мальтазы оказалась нормальной. Резко снижена была активность гексокиназы в эритроцитах.
Дифференциальный диагноз у новорожденных проводится с сифилисом (смотри полный свод знаний), токсоплазмозом (смотри полный свод знаний), цитомегалией (смотри полный свод знаний), заболеваниями печени, в более старшем возрасте — с болезнями Гоше (смотри полный свод знаний Гоше болезнь), Ниманна—Пика (смотри полный свод знаний Ниманна—Пика болезнь), опухолями печени, миотонией (смотри полный свод знаний), ксантоматозом (смотри полный свод знаний).
Нервно-мышечные нарушения могут имитировать прогрессирующую мышечную дистрофию, невральную амиотрофию Шарко—Мари, спинальную амиотрофию Верднига—Гоффманна (при генерализованном Гликогеноз II типа), в связи с чем необходимо проведение соответствующих биохимический и морфологически исследований в каждом случае поражения мышц.
Лечение
Специфического лечения нет. Патогенетическая терапия направлена на борьбу с ацидозом (смотри полный свод знаний), кетозом. В некоторых случаях эффективно применение глюкагона, анаболических и стероидных гормонов. Частые приёмы пищи с высоким содержанием легко усвояемых углеводов необходимы при гипогликемическом синдроме. Имеются попытки введения больным недостающих энзимов.
При мышечных Гликогеноз улучшение отмечается после назначения фруктозы внутрь по 50,0—100,0 грамм в день, витаминов, АТФ. Делаются попытки хирургического лечения I и III типов Гликогеноз (портокавальная транспозиция сосудов, перевязка портальной вены и наложение анастомоза конец в бок — V. portae и v. cava inf.).
Смотри также Энзимопатии.
|
Бадалян Л.О.; Князев Ю.А.; Серов В.В. |

|
⇐ Перейти на главную страницу сайта |
⇑ Вернуться в начало страницы ⇑ |
Библиотека Ordo Deus ⇒ |
⇐ Глаукома |
⇓ Полный свод знаний. Том первый А. ⇓ |
Глиобластома ⇒ |
|
Все статьи в полном изложении, Вы можете найти в большой медицинской энциклопедии — Главный редактор: академик АН СССР (РАН) и АМН СССР (РАМН) Б.В. Петровский. — Москва издательство «Советская энциклопедия» 1989г. |
|
Внимание! Вы находитесь в библиотеке «Ordo Deus». Все книги в электронном варианте, содержащиеся в библиотеке «Ordo Deus», принадлежат их законным владельцам (авторам, переводчикам, издательствам). Все книги и статьи взяты из открытых источников и размещаются здесь только для чтения. |
|
Библиотека «Ordo Deus» не преследует никакой коммерческой выгоды. |
|
Все авторские права сохраняются за правообладателями. Если Вы являетесь автором данного документа и хотите дополнить его или изменить, уточнить реквизиты автора, опубликовать другие документы или возможно вы не желаете, чтобы какой-то из ваших материалов находился в библиотеке, пожалуйста, свяжитесь с нами по e-mail: |
Вас категорически не устраивает перспектива безвозвратно исчезнуть из этого мира? Вы не желаете закончить свой жизненный путь в виде омерзительной гниющей органической массы пожираемой копошащимися в ней могильными червями? Вы желаете вернувшись в молодость прожить ещё одну жизнь? Начать всё заново? Исправить совершённые ошибки? Осуществить несбывшиеся мечты? Перейдите по ссылке: «главная страница».
|
© Ordo Deus, 2010. При копировании ссылка на сайт http://www.ordodeus.ru обязательна. |