Лоренса — Муна — Бидля синдром |
||
 |
 |
|
Оглавление
|
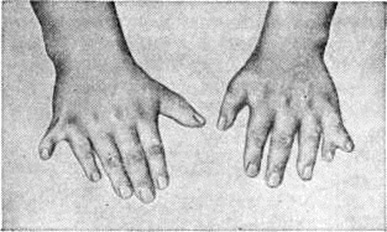
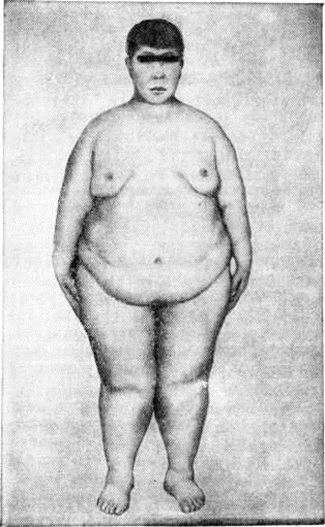
Лоренса — Муна — Бидля синдромЛоренса — Муна — Бидля синдром (J. Z. Laurence, английский офтальмолог, 1830—1874; R. Ch. Moon, американский офтальмолог, 1844— 1914; A. Biedl, чешский терапевт, 1869—1933) — нейроэндокринное заболевание, проявляющееся пигментным ретинитом, ожирением, полидактилией, гипогенитализмом и умственной отсталостью. Синдром описан в 1866 год Лоренсом и Муном как сочетание пигментного ретинита с гипогенитализмом, задержкой роста и олигофренией. В 1920 год Барде (G. Bardet) обратил внимание на полидактилию при этом синдроме, а в 1922 год Бидль описал другие пороки развития при этом синдроме. Описано немногим более 400 больных. Заболевание часто имеет семейный характер, чаще встречается у лиц мужского пола. Этиология и патогенез изучены недостаточно. Наибольшее значение придаётся генетическим факторам, однако тип наследования не уточнён. Допускается, что синдром является следствием внутриутробного повреждения плода, например, при токсоплазмозе (смотри полный свод знаний), краснухе (смотри полный свод знаний) у беременных. Наряду с врождёнными пороками развития скелета, глаз, мозга, внутренних органов, прогрессирующими дистрофическими изменениями (например, сетчатки глаз, почек) придаётся значение вторичным расстройствам, связанным с нарушением функции гипоталамических центров. Патологическая анатомия. Морфологически изменения в мозге не специфичны для Лоренса — Муна — Бидля синдром и у многих больных вообще отсутствуют. Описаны дистрофические изменения ядер гипоталамуса с уменьшением числа ганглиозных клеток и замещение их глиозными элементами, а также атрофия мозговых извилин, врождённое отсутствие мозолистого тела и другие Часто наблюдаются дефекты развития почек — фетальная дольчатость, гипоплазия, дисплазия; микроскопически в почках обнаруживается широкий спектр изменений — от небольшого разрастания мезенхимальных элементов до выраженной мезангиальной пролиферации, кистозного расширения канальцев с образованием кортикальных и медуллярных кист, перигломерулярный и интерстициальный фиброз, хронический воспалительная клеточная инфильтрация. Врождённые пороки сердца и сосудов выявлены при аутопсии у 2/5 умерших. Половые органы, как правило, недоразвиты. Клиническая картина. Ожирение (смотри полный свод знаний) встречается у 81—95% больных, чаще начинается уже на первом году жизни и увеличивается с возрастом. Пигментная дистрофия сетчатки, или пигментный ретинит (смотри полный свод знаний Тапеторетиналъные дистрофии), хотя и относится к кардинальным симптомам заболевания, описана лишь у 15% больных; нарушения зрения наблюдаются у 92—93% больных (более 70% больных слепнут). Причиной прогрессирующей потери зрения наряду с пигментным ретинитом являются атрофия зрительного нерва (смотри полный свод знаний), глаукома (смотри полный свод знаний), катаракта (смотри полный свод знаний), близорукость (смотри полный свод знаний); описаны пороки развития глаз: микрофтальмия (смотри полный свод знаний Глаз), анофтальмия (смотри полный свод знаний), аниридия (смотри полный свод знаний), колобома радужной оболочки (смотри полный свод знаний Колобома). Полидактилия, обычно шестипалость (рисунок 1), встречается у 70—80% больных; у некоторых больных имеется синдактилия (смотри полный свод знаний), иногда в сочетании с полидактилией, брахидактилия (смотри полный свод знаний Кисть), плоскостопие (смотри полный свод знаний). Описаны пороки развития черепа: микроцефалия (смотри полный свод знаний), гидроцефалия (смотри полный свод знаний), брахицефалия (смотри полный свод знаний), лобный гиперостоз (смотри полный свод знаний Морганьи синдром), деформация турецкого седла, асимметрия лица, а также дефекты позвонков и рёбер. Больные обычно малого роста, созревание скелета замедлено. Гипогонадизм (смотри полный свод знаний) наблюдается у 74—85% больных мужчин и у 45—53% женщин; он может быть связан как с первичной недостаточностью половых желёз, так и с понижением продукции гонадотропинов. У мужчин отмечается характерный евнухоидный вид, резкое ожирение, нередко гинекомастия (рисунок 2), малые размеры яичек и наружных половых органов, рост волос на лобке по женскому типу. У женщин могут полностью отсутствовать вторичные половые признаки, наблюдаться аменорея и гипоплазия половых органов, вместе с тем в ряде случаев возможно нормальное половое развитие со способностью к деторождению. |
У некоторых больных имелись нарушения толерантности к глюкозе вплоть до развития сахарного диабета; иногда развивается несахарный диабет, отмечается артериальная гипертензия.
Расстройства психики имеют место у 70—85% больных; у некоторых олигофрения (смотри полный свод знаний) с раннего детства, но описано внезапное нарушение интеллекта (с 7—8-летнего возраста). Степень изменения психики различна. Отмечаются неврологический расстройства, на ЭЭГ часто наблюдается нарушение регулярности основных ритмов, диффузная дизритмия; изредка— экстрапирамидные нарушения: спастические параличи конечностей, гипо и гиперрефлексия.
 |
|  |
Рис. 1. | ||
|
| |
Рис. 2. | ||
Врождённые пороки сердца, дефекты развития аорты и коронарных сосудов при жизни диагностируются редко.
На фоне врождённых дефектов почек (поликистоз, гипоплазия, гломерулярная дисплазия) возникают воспалительные процессы (хронический гломерулонефрит, пиелонефрит, абсцессы почек), которые обнаруживаются при урологический обследовании с использованием радиорентгенологическое методов и биопсии почек.
Клейн и Амманн (D. Klein, F. Ammann, 1969) предложили выделять полную форму синдрома (все пять кардинальных симптомов), неполную (один или два симптома отсутствуют), абортивную (один-два симптома или нечёткие проявления всех), атипичную (пигментного ретинита нет, но отмечаются другие поражения глаз) и экстенсивную форму (наряду с пятью основными симптомами имеются другие пороки развития). Полная форма встречается относительно редко: из 132 случаев, собранных по литературе Теленом (Е. Thelen, 1958), она выявлена лишь в 26.
Диагноз при полной форме синдрома не представляет затруднений. Полидактилия выявляется уже при рождении. Нередко на первом году жизни развивается ожирение. В дальнейшем выявляются другие симптомы. Дифференциальный диагноз проводится с адипозо-генитальной дистрофией (смотри полный свод знаний), которой не свойственны поражения глаз, полидактилия и другие пороки развития, олигофрения, а также с синдромом Альстрема — Халлгрена, характеризующимся сочетанием пигментного ретинита с ожирением, глухотой, сахарным диабетом (иногда психические расстройства) при отсутствии полидактилии и гипогонадизма.
Лечение симптоматическое.
Прогноз неблагоприятный. Обычно больные слепнут, ожирение и почечная недостаточность усиливаются. Больные чаще умирают в молодом возрасте; старшей из умерших было 50 лет. У трети умерших причиной смерти была уремия.
|
Раскин А.М. |

|
⇐ Перейти на главную страницу сайта |
⇑ Вернуться в начало страницы ⇑ |
Библиотека Ordo Deus ⇒ |
⇐ Лордоз |
⇓ Полный свод знаний. Том первый А. ⇓ |
Лоу синдром ⇒ |
|
Все статьи в полном изложении, Вы можете найти в большой медицинской энциклопедии — Главный редактор: академик АН СССР (РАН) и АМН СССР (РАМН) Б.В. Петровский. — Москва издательство «Советская энциклопедия» 1989г. |
|
Внимание! Вы находитесь в библиотеке «Ordo Deus». Все книги в электронном варианте, содержащиеся в библиотеке «Ordo Deus», принадлежат их законным владельцам (авторам, переводчикам, издательствам). Все книги и статьи взяты из открытых источников и размещаются здесь только для чтения. |
|
Вся информация на сайте Ordo Deus находится в свободном доступе. Ordo Deus не предоставляет информацию на платной основе. |
|
Все авторские права сохраняются за правообладателями. Если Вы являетесь автором данного документа и хотите дополнить его или изменить, уточнить реквизиты автора, опубликовать другие документы или возможно вы не желаете, чтобы какой-то из ваших материалов находился в библиотеке, пожалуйста, свяжитесь с нами по e-mail: |
Вас категорически не устраивает перспектива безвозвратно исчезнуть из этого мира? Вы не желаете закончить свой жизненный путь в виде омерзительной гниющей органической массы пожираемой копошащимися в ней могильными червями? Вы желаете вернувшись в молодость прожить ещё одну жизнь? Начать всё заново? Исправить совершённые ошибки? Осуществить несбывшиеся мечты? Перейдите по ссылке: «главная страница».
|
© Ordo Deus, 2010. При копировании ссылка на сайт http://www.ordodeus.ru обязательна. |